Introduction to GenomicState
Leonardo Collado-Torres
Lieber Institute for Brain Development, Johns Hopkins Medical CampusCenter for Computational Biology, Johns Hopkins Universitylcolladotor@gmail.com
12 December 2024
Source:vignettes/GenomicState.Rmd
GenomicState.RmdBasics
Install GenomicState
R is an open-source statistical environment which can be
easily modified to enhance its functionality via packages. GenomicState
is a R package available via Bioconductor. R
can be installed on any operating system from CRAN after which you can install
GenomicState
by using the following commands in your R session:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("GenomicState")
## Check that you have a valid Bioconductor installation
BiocManager::valid()Required knowledge
GenomicState is based on many other packages and in particular in those that have implemented the infrastructure needed for dealing with annotation data. That is, packages like rtracklayer that allow you to import the data. A GenomicState user is not expected to deal with those packages directly but will need to be familiar with derfinder and derfinderPlot to understand the results GenomicState generates. Furthermore, it’ll be useful for the user to know the syntax of AnnotationHub (Morgan and Shepherd, 2024) in order to query and load the data provided by this package.
If you are asking yourself the question “Where do I start using Bioconductor?” you might be interested in this blog post.
Asking for help
As package developers, we try to explain clearly how to use our
packages and in which order to use the functions. But R and
Bioconductor have a steep learning curve so it is critical
to learn where to ask for help. The blog post quoted above mentions some
but we would like to highlight the Bioconductor support site
as the main resource for getting help regarding Bioconductor. Other
alternatives are available such as creating GitHub issues and tweeting.
However, please note that if you want to receive help you should adhere
to the posting
guidelines. It is particularly critical that you provide a small
reproducible example and your session information so package developers
can track down the source of the error.
Citing GenomicState
We hope that GenomicState will be useful for your research. Please use the following information to cite the package and the overall approach. Thank you!
## Citation info
citation("GenomicState")
#> To cite package 'GenomicState' in publications use:
#>
#> Collado-Torres L (2024). _Build and access GenomicState objects for
#> use with derfinder tools from sources like Gencode_.
#> doi:10.18129/B9.bioc.GenomicState
#> <https://doi.org/10.18129/B9.bioc.GenomicState>,
#> https://github.com/LieberInstitute/GenomicState - R package version
#> 0.99.16, <http://www.bioconductor.org/packages/GenomicState>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Manual{,
#> title = {Build and access GenomicState objects for use with derfinder tools from sources like Gencode},
#> author = {Leonardo Collado-Torres},
#> year = {2024},
#> url = {http://www.bioconductor.org/packages/GenomicState},
#> note = {https://github.com/LieberInstitute/GenomicState - R package version 0.99.16},
#> doi = {10.18129/B9.bioc.GenomicState},
#> }Overview
The GenomicState
package was developed for speeding up analyses that require these
objects and in particular those that rely on Gencode annotation data.
The package GenomicState
provides functions for building GenomicState objects from
diverse annotation sources such as Gencode.
It also provides a way to load pre-computed GenomicState
objects if you are working at JHPCE. These GenomicState
objects are normally created using derfinder::makeGenomicState()
and can be used for annotating regions with derfinder::annotateRegions()
which are in turn used by derfinderPlot::plotRegionCoverage().
To get started, load the GenomicState package.
AnnotationHub
Using the GencodeStateHub() function you can query and
download the data from GenomicState
using AnnotationHub
(Morgan and Shepherd, 2024).
## Query AnnotationHub for the GenomicState object for Gencode v31 on
## hg19 coordinates
hub_query_gs_gencode_v31_hg19 <- GenomicStateHub(
version = "31",
genome = "hg19",
filetype = "GenomicState"
)
hub_query_gs_gencode_v31_hg19
#> AnnotationHub with 1 record
#> # snapshotDate(): 2024-10-28
#> # names(): AH75184
#> # $dataprovider: GENCODE
#> # $species: Homo sapiens
#> # $rdataclass: list
#> # $rdatadateadded: 2019-10-22
#> # $title: GenomicState for Gencode v31 on hg19 coordinates
#> # $description: Gencode v31 GenomicState from derfinder::makeGenomicState() ...
#> # $taxonomyid: 9606
#> # $genome: GRCh37
#> # $sourcetype: GTF
#> # $sourceurl: ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/releas...
#> # $sourcesize: NA
#> # $tags: c("Gencode", "GenomicState", "hg19", "v31")
#> # retrieve record with 'object[["AH75184"]]'
## Check the metadata
mcols(hub_query_gs_gencode_v31_hg19)
#> DataFrame with 1 row and 15 columns
#> title dataprovider species taxonomyid genome
#> <character> <character> <character> <integer> <character>
#> AH75184 GenomicState for Gen.. GENCODE Homo sapiens 9606 GRCh37
#> description coordinate_1_based maintainer
#> <character> <integer> <character>
#> AH75184 Gencode v31 GenomicS.. 1 Leonardo Collado-Tor..
#> rdatadateadded preparerclass tags rdataclass
#> <character> <character> <AsIs> <character>
#> AH75184 2019-10-22 GenomicState Gencode,GenomicState,hg19,... list
#> rdatapath sourceurl sourcetype
#> <character> <character> <character>
#> AH75184 GenomicState/gencode.. ftp://ftp.ebi.ac.uk/.. GTF
## Access the file through AnnotationHub
if (length(hub_query_gs_gencode_v31_hg19) == 1) {
hub_gs_gencode_v31_hg19 <- hub_query_gs_gencode_v31_hg19[[1]]
hub_gs_gencode_v31_hg19
}
#> loading from cache
#> $fullGenome
#> GRanges object with 659263 ranges and 5 metadata columns:
#> seqnames ranges strand | theRegion tx_id
#> <Rle> <IRanges> <Rle> | <character> <IntegerList>
#> 1 chr1 11869-12227 + | exon 1,2
#> 2 chr1 12228-12612 + | intron 1,2
#> 3 chr1 12613-12721 + | exon 1,2
#> 4 chr1 12722-12974 + | intron 1,2
#> 5 chr1 12975-13052 + | exon 2
#> ... ... ... ... . ... ...
#> 659259 chrY 59208555-59214013 * | intergenic
#> 659260 chrY 59276440-59311662 * | intergenic
#> 659261 chrY 59311997-59318040 * | intergenic
#> 659262 chrY 59318921-59330251 * | intergenic
#> 659263 chrY 59360549-59373566 * | intergenic
#> tx_name gene symbol
#> <CharacterList> <IntegerList> <CharacterList>
#> 1 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> 2 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> 3 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> 4 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> 5 ENST00000450305.2_1 26085 DDX11L1
#> ... ... ... ...
#> 659259
#> 659260
#> 659261
#> 659262
#> 659263
#> -------
#> seqinfo: 24 sequences from hg19 genome
#>
#> $codingGenome
#> GRanges object with 878954 ranges and 5 metadata columns:
#> seqnames ranges strand | theRegion tx_id
#> <Rle> <IRanges> <Rle> | <character> <IntegerList>
#> 1 chr1 9869-11868 + | promoter 1,2
#> 2 chr1 11869-12227 + | exon 1,2
#> 3 chr1 12228-12612 + | intron 1,2
#> 4 chr1 12613-12721 + | exon 1,2
#> 5 chr1 12722-12974 + | intron 1,2
#> ... ... ... ... . ... ...
#> 878950 chrY 59208555-59212013 * | intergenic
#> 878951 chrY 59276440-59311662 * | intergenic
#> 878952 chrY 59313997-59318040 * | intergenic
#> 878953 chrY 59320921-59328251 * | intergenic
#> 878954 chrY 59362549-59373566 * | intergenic
#> tx_name gene symbol
#> <CharacterList> <IntegerList> <CharacterList>
#> 1 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> 2 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> 3 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> 4 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> 5 ENST00000450305.2_1,ENST00000456328.2_1 26085 DDX11L1
#> ... ... ... ...
#> 878950
#> 878951
#> 878952
#> 878953
#> 878954
#> -------
#> seqinfo: 24 sequences from hg19 genomeUsing the objects
To show how we can use these objects, first we build those for Gencode version 31 on hg19 coordinates.
## Load the example TxDb object
## or start from scratch with:
## txdb_v31_hg19_chr21 <- gencode_txdb(version = '31', genome = 'hg19',
## chrs = 'chr21')
txdb_v31_hg19_chr21 <- AnnotationDbi::loadDb(
system.file("extdata", "txdb_v31_hg19_chr21.sqlite",
package = "GenomicState"
)
)
#> Loading required package: GenomicFeatures
#> Loading required package: S4Vectors
#> Loading required package: stats4
#>
#> Attaching package: 'S4Vectors'
#> The following object is masked from 'package:utils':
#>
#> findMatches
#> The following objects are masked from 'package:base':
#>
#> expand.grid, I, unname
#> Loading required package: IRanges
#> Loading required package: GenomeInfoDb
#> Loading required package: GenomicRanges
#> Loading required package: AnnotationDbi
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#>
#> Attaching package: 'Biobase'
#> The following object is masked from 'package:AnnotationHub':
#>
#> cache
## Build the GenomicState and annotated genes
genes_v31_hg19_chr21 <- gencode_annotated_genes(txdb_v31_hg19_chr21)
#> 2024-12-12 22:30:33.505134 annotating the transcripts
#> No annotationPackage supplied. Trying org.Hs.eg.db.
#> Loading required package: org.Hs.eg.db
#>
#> Getting TSS and TSE.
#> Getting CSS and CSE.
#> Getting exons.
#> Annotating genes.
#> 'select()' returned 1:many mapping between keys and columns
gs_v31_hg19_chr21 <- gencode_genomic_state(txdb_v31_hg19_chr21)
#> 2024-12-12 22:30:55.884377 making the GenomicState object
#> extendedMapSeqlevels: sequence names mapped from NCBI to UCSC for species homo_sapiens
#> 'select()' returned 1:1 mapping between keys and columns
#> 2024-12-12 22:31:01.676907 finding gene symbols
#> 'select()' returned 1:many mapping between keys and columns
#> 2024-12-12 22:31:01.958252 adding gene symbols to the GenomicStateYou can alternatively use the files hosted in AnnotationHub (Morgan and Shepherd, 2024) which will be faster in general.
## Create the AnnotationHub object once and re-use it to speed up things
ah <- AnnotationHub::AnnotationHub()
## Find the TxDb object for hg19 Gencode version 31
hub_query_txdb_gencode_v31_hg19 <- GenomicStateHub(
version = "31",
genome = "hg19",
filetype = "TxDb", ah = ah
)
hub_query_txdb_gencode_v31_hg19
#> AnnotationHub with 1 record
#> # snapshotDate(): 2024-10-28
#> # names(): AH75182
#> # $dataprovider: GENCODE
#> # $species: Homo sapiens
#> # $rdataclass: TxDb
#> # $rdatadateadded: 2019-10-22
#> # $title: TxDb for Gencode v31 on hg19 coordinates
#> # $description: Gencode v31 TxDb object on hg19 coordinates. This is useful ...
#> # $taxonomyid: 9606
#> # $genome: GRCh37
#> # $sourcetype: GTF
#> # $sourceurl: ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/releas...
#> # $sourcesize: NA
#> # $tags: c("Gencode", "GenomicState", "hg19", "v31")
#> # retrieve record with 'object[["AH75182"]]'
## Now the Annotated Genes for hg19 Gencode v31
hub_query_genes_gencode_v31_hg19 <- GenomicStateHub(
version = "31",
genome = "hg19",
filetype = "AnnotatedGenes", ah = ah
)
hub_query_genes_gencode_v31_hg19
#> AnnotationHub with 1 record
#> # snapshotDate(): 2024-10-28
#> # names(): AH75183
#> # $dataprovider: GENCODE
#> # $species: Homo sapiens
#> # $rdataclass: GRanges
#> # $rdatadateadded: 2019-10-22
#> # $title: Annotated genes for Gencode v31 on hg19 coordinates
#> # $description: Gencode v31 annotated genes from bumphunter::annotateTranscr...
#> # $taxonomyid: 9606
#> # $genome: GRCh37
#> # $sourcetype: GTF
#> # $sourceurl: ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/releas...
#> # $sourcesize: NA
#> # $tags: c("Gencode", "GenomicState", "hg19", "v31")
#> # retrieve record with 'object[["AH75183"]]'
## And finally the GenomicState for hg19 Gencode v31
hub_query_gs_gencode_v31_hg19 <- GenomicStateHub(
version = "31",
genome = "hg19",
filetype = "GenomicState", ah = ah
)
hub_query_gs_gencode_v31_hg19
#> AnnotationHub with 1 record
#> # snapshotDate(): 2024-10-28
#> # names(): AH75184
#> # $dataprovider: GENCODE
#> # $species: Homo sapiens
#> # $rdataclass: list
#> # $rdatadateadded: 2019-10-22
#> # $title: GenomicState for Gencode v31 on hg19 coordinates
#> # $description: Gencode v31 GenomicState from derfinder::makeGenomicState() ...
#> # $taxonomyid: 9606
#> # $genome: GRCh37
#> # $sourcetype: GTF
#> # $sourceurl: ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/releas...
#> # $sourcesize: NA
#> # $tags: c("Gencode", "GenomicState", "hg19", "v31")
#> # retrieve record with 'object[["AH75184"]]'
## If you want to access the files use the double bracket AnnotationHub syntax
## to retrieve the R objects from the web.
if (FALSE) {
hub_txdb_gencode_v31_hg19 <- hub_query_txdb_gencode_v31_hg19[[1]]
hub_genes_gencode_v31_hg19 <- hub_query_genes_gencode_v31_hg19[[1]]
hub_gs_gencode_v31_hg19 <- hub_query_gs_gencode_v31_hg19[[1]]
}Next we load a series of related packages that use the objects we created with GenomicState or downloaded from AnnotationHub (Morgan and Shepherd, 2024).
## Load external packages
library("derfinder")
library("derfinderPlot")
#> Registered S3 method overwritten by 'GGally':
#> method from
#> +.gg ggplot2
library("bumphunter")
#> Loading required package: foreach
#> Loading required package: iterators
#> Loading required package: parallel
#> Loading required package: locfit
#> locfit 1.5-9.10 2024-06-24
library("GenomicRanges")Next we can prepare the needed for running
derfinderPlot::plotRegionCoverage() where we use the
TxDb object, the GenomicState and the
annotated genes we prepared for Gencode v31 on hg19.
## Some example regions from derfinder (set the chromosome lengths)
regions <- genomeRegions$regions[1:2]
seqlengths(regions) <- seqlengths(txdb_v31_hg19_chr21)[
names(seqlengths(regions))
]
## Annotate them
nearestAnnotation <- matchGenes(x = regions, subject = genes_v31_hg19_chr21)
annotatedRegions <- annotateRegions(
regions = regions,
genomicState = gs_v31_hg19_chr21$fullGenome, minoverlap = 1
)
#> 2024-12-12 22:31:12.075855 annotateRegions: counting
#> 2024-12-12 22:31:12.135269 annotateRegions: annotating
## Obtain fullCov object
fullCov <- list("chr21" = genomeDataRaw$coverage)
regionCov <- getRegionCoverage(fullCov = fullCov, regions = regions)
#> 2024-12-12 22:31:12.230014 getRegionCoverage: processing chr21
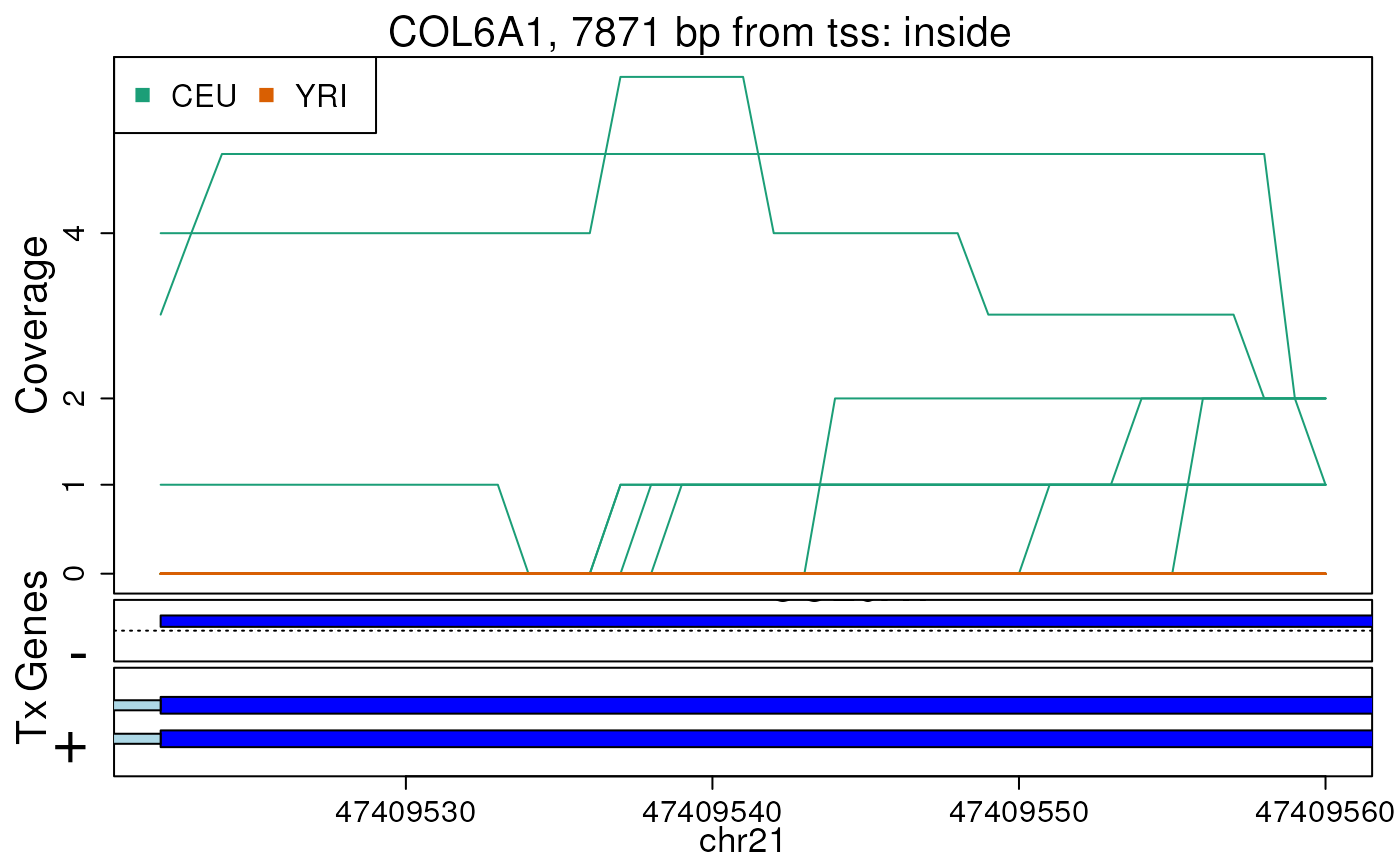
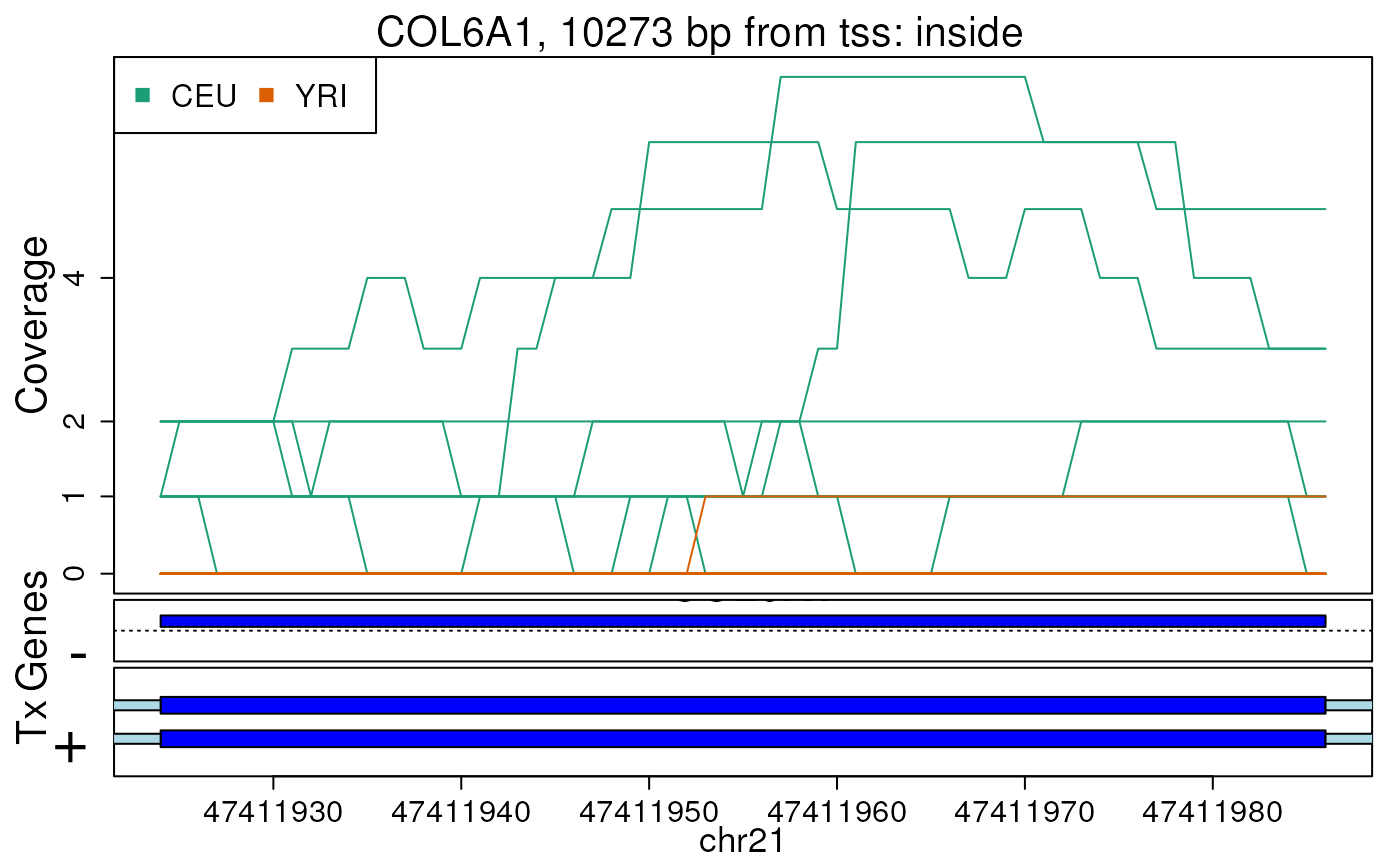
#> 2024-12-12 22:31:12.268195 getRegionCoverage: done processing chr21And now we can make the example plot as shown below.
## now make the plot
plotRegionCoverage(
regions = regions, regionCoverage = regionCov,
groupInfo = genomeInfo$pop, nearestAnnotation = nearestAnnotation,
annotatedRegions = annotatedRegions, whichRegions = 1:2,
txdb = txdb_v31_hg19_chr21, verbose = FALSE
)
JHPCE
You can also access the data locally using the function
local_metadata() which works at JHPCE or anywhere where you have
re-created the files from this package. This returns a
data.frame() which you can subset. It also inclused the R
code for loading the data which you can do using
eval(parse(text = local_metadata()$loadCode)) as shown
below.
## Get the local metadata
meta <- local_metadata()
## Subset to the data of interest, lets say hg19 TxDb for v31
interest <- subset(meta, RDataClass == "TxDb" & Tags == "Gencode:v31:hg19")
## Next you can load the data
if (file.exists(interest$RDataPath)) {
## This only works at JHPCE
eval(parse(text = interest$loadCode))
## Explore the loaded object (would be gencode_v31_hg19_txdb in this case)
gencode_v31_hg19_txdb
}Build objects
The objects provided by GenomicState through AnnotationHub
(Morgan and Shepherd, 2024) were built using code like the one included
below which is how the Gencode version 23 for hg19 files were built.
outdir <- "gencode"
dir.create(outdir, showWarnings = FALSE)
## Build and save the TxDb object
gencode_v23_hg19_txdb <- gencode_txdb("23", "hg19")
saveDb(gencode_v23_hg19_txdb,
file = file.path(outdir, "gencode_v23_hg19_txdb.sqlite")
)
## Build and save the annotateTranscripts output
gencode_v23_hg19_annotated_genes <- gencode_annotated_genes(
gencode_v23_hg19_txdb
)
save(gencode_v23_hg19_annotated_genes,
file = file.path(outdir, "gencode_v23_hg19_annotated_genes.rda")
)
## Build and save the GenomicState
gencode_v23_hg19_GenomicState <- gencode_genomic_state(
gencode_v23_hg19_txdb
)
save(gencode_v23_hg19_GenomicState,
file = file.path(outdir, "gencode_v23_hg19_GenomicState.rda")
)For more details check the source files:
## R commands for building the files:
system.file("scripts", "make-data_gencode_human.R",
package = "GenomicState"
)
#> [1] "/__w/_temp/Library/GenomicState/scripts/make-data_gencode_human.R"
## The above file was created by this one:
system.file("scripts", "generate_make_data_gencode_human.R",
package = "GenomicState"
)
#> [1] "/__w/_temp/Library/GenomicState/scripts/generate_make_data_gencode_human.R"Reproducibility
The GenomicState package (Collado-Torres, 2024) was made possible thanks to:
- R (R Core Team, 2024)
- BiocStyle (Oleś, 2024)
- GenomeInfoDb (Arora, Morgan, Carlson, and Pagès, 2017)
- knitr (Xie, 2014)
- RefManageR (McLean, 2017)
- rmarkdown (Allaire, Xie, Dervieux, McPherson, Luraschi, Ushey, Atkins, Wickham, Cheng, Chang, and Iannone, 2024)
- rtracklayer (Lawrence, Gentleman, and Carey, 2009)
- sessioninfo (Wickham, Chang, Flight, Müller, and Hester, 2021)
- testthat (Wickham, 2011)
- GenomicFeatures (Lawrence, Huber, Pagès, Aboyoun, Carlson, Gentleman, Morgan, and Carey, 2013)
- bumphunter (Jaffe, Murakami, Lee, Leek, Fallin, Feinberg, and Irizarry, 2012)
- derfinder (Collado-Torres, Nellore, Frazee, Wilks, Love, Langmead, Irizarry, Leek, and Jaffe, 2017)
- AnnotationDbi (Pagès, Carlson, Falcon, and Li, 2024)
- IRanges (Lawrence, Huber, Pagès et al., 2013)
- org.Hs.eg.db (Carlson, 2024)
- glue (Hester and Bryan, 2024)
- AnnotationHub (Morgan and Shepherd, 2024)
- AnnotationHubData (Bioconductor Package Maintainer, 2024)
- GenomicRanges (Lawrence, Huber, Pagès et al., 2013)
- txdbmaker (Pagès, Carlson, Aboyoun, Falcon, and Morgan, 2024)
Code for creating the vignette
## Create the vignette
library("rmarkdown")
system.time(render("GenomicState.Rmd"))
## Extract the R code
library("knitr")
knit("GenomicState.Rmd", tangle = TRUE)Date the vignette was generated.
#> [1] "2024-12-12 22:31:13 UTC"Wallclock time spent generating the vignette.
#> Time difference of 59.052 secsR session information.
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
#> version R version 4.4.2 (2024-10-31)
#> os Ubuntu 24.04.1 LTS
#> system x86_64, linux-gnu
#> ui X11
#> language en
#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
#> date 2024-12-12
#> pandoc 3.5 @ /usr/bin/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
#> abind 1.4-8 2024-09-12 [1] RSPM (R 4.4.0)
#> AnnotationDbi * 1.68.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> AnnotationFilter 1.30.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> AnnotationHub * 3.14.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> backports 1.5.0 2024-05-23 [1] RSPM (R 4.4.0)
#> base64enc 0.1-3 2015-07-28 [2] RSPM (R 4.4.0)
#> bibtex 0.5.1 2023-01-26 [1] RSPM (R 4.4.0)
#> Biobase * 2.66.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> BiocFileCache * 2.14.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> BiocGenerics * 0.52.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> BiocIO 1.16.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> BiocManager 1.30.25 2024-08-28 [2] CRAN (R 4.4.2)
#> BiocParallel 1.40.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> BiocStyle * 2.34.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> BiocVersion 3.20.0 2024-10-21 [2] Bioconductor 3.20 (R 4.4.2)
#> biomaRt 2.62.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> Biostrings 2.74.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> biovizBase 1.54.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> bit 4.5.0.1 2024-12-03 [1] RSPM (R 4.4.0)
#> bit64 4.5.2 2024-09-22 [1] RSPM (R 4.4.0)
#> bitops 1.0-9 2024-10-03 [1] RSPM (R 4.4.0)
#> blob 1.2.4 2023-03-17 [1] RSPM (R 4.4.0)
#> bookdown 0.41 2024-10-16 [1] RSPM (R 4.4.0)
#> BSgenome 1.74.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> bslib 0.8.0 2024-07-29 [2] RSPM (R 4.4.0)
#> bumphunter * 1.48.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> cachem 1.1.0 2024-05-16 [2] RSPM (R 4.4.0)
#> checkmate 2.3.2 2024-07-29 [1] RSPM (R 4.4.0)
#> cli 3.6.3 2024-06-21 [2] RSPM (R 4.4.0)
#> cluster 2.1.8 2024-12-11 [3] RSPM (R 4.4.0)
#> codetools 0.2-20 2024-03-31 [3] CRAN (R 4.4.2)
#> colorspace 2.1-1 2024-07-26 [1] RSPM (R 4.4.0)
#> crayon 1.5.3 2024-06-20 [2] RSPM (R 4.4.0)
#> curl 6.0.1 2024-11-14 [2] RSPM (R 4.4.0)
#> data.table 1.16.4 2024-12-06 [1] RSPM (R 4.4.0)
#> DBI 1.2.3 2024-06-02 [1] RSPM (R 4.4.0)
#> dbplyr * 2.5.0 2024-03-19 [1] RSPM (R 4.4.0)
#> DelayedArray 0.32.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> derfinder * 1.40.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> derfinderHelper 1.40.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> derfinderPlot * 1.40.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> desc 1.4.3 2023-12-10 [2] RSPM (R 4.4.0)
#> dichromat 2.0-0.1 2022-05-02 [1] RSPM (R 4.4.0)
#> digest 0.6.37 2024-08-19 [2] RSPM (R 4.4.0)
#> doRNG 1.8.6 2023-01-16 [1] RSPM (R 4.4.0)
#> dplyr 1.1.4 2023-11-17 [1] RSPM (R 4.4.0)
#> ensembldb 2.30.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> evaluate 1.0.1 2024-10-10 [2] RSPM (R 4.4.0)
#> fansi 1.0.6 2023-12-08 [2] RSPM (R 4.4.0)
#> fastmap 1.2.0 2024-05-15 [2] RSPM (R 4.4.0)
#> filelock 1.0.3 2023-12-11 [1] RSPM (R 4.4.0)
#> foreach * 1.5.2 2022-02-02 [1] RSPM (R 4.4.0)
#> foreign 0.8-87 2024-06-26 [3] CRAN (R 4.4.2)
#> Formula 1.2-5 2023-02-24 [1] RSPM (R 4.4.0)
#> fs 1.6.5 2024-10-30 [2] RSPM (R 4.4.0)
#> generics 0.1.3 2022-07-05 [1] RSPM (R 4.4.0)
#> GenomeInfoDb * 1.42.1 2024-11-28 [1] Bioconductor 3.20 (R 4.4.2)
#> GenomeInfoDbData 1.2.13 2024-12-12 [1] Bioconductor
#> GenomicAlignments 1.42.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> GenomicFeatures * 1.58.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> GenomicFiles 1.42.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> GenomicRanges * 1.58.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> GenomicState * 0.99.16 2024-12-12 [1] Bioconductor
#> GGally 2.2.1 2024-02-14 [1] RSPM (R 4.4.0)
#> ggbio 1.54.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> ggplot2 3.5.1 2024-04-23 [1] RSPM (R 4.4.0)
#> ggstats 0.7.0 2024-09-22 [1] RSPM (R 4.4.0)
#> glue 1.8.0 2024-09-30 [2] RSPM (R 4.4.0)
#> graph 1.84.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> gridExtra 2.3 2017-09-09 [1] RSPM (R 4.4.0)
#> gtable 0.3.6 2024-10-25 [1] RSPM (R 4.4.0)
#> Hmisc 5.2-1 2024-12-02 [1] RSPM (R 4.4.0)
#> hms 1.1.3 2023-03-21 [1] RSPM (R 4.4.0)
#> htmlTable 2.4.3 2024-07-21 [1] RSPM (R 4.4.0)
#> htmltools 0.5.8.1 2024-04-04 [2] RSPM (R 4.4.0)
#> htmlwidgets 1.6.4 2023-12-06 [2] RSPM (R 4.4.0)
#> httr 1.4.7 2023-08-15 [1] RSPM (R 4.4.0)
#> httr2 1.0.7 2024-11-26 [2] RSPM (R 4.4.0)
#> IRanges * 2.40.1 2024-12-05 [1] Bioconductor 3.20 (R 4.4.2)
#> iterators * 1.0.14 2022-02-05 [1] RSPM (R 4.4.0)
#> jquerylib 0.1.4 2021-04-26 [2] RSPM (R 4.4.0)
#> jsonlite 1.8.9 2024-09-20 [2] RSPM (R 4.4.0)
#> KEGGREST 1.46.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> knitr 1.49 2024-11-08 [2] RSPM (R 4.4.0)
#> lattice 0.22-6 2024-03-20 [3] CRAN (R 4.4.2)
#> lazyeval 0.2.2 2019-03-15 [1] RSPM (R 4.4.0)
#> lifecycle 1.0.4 2023-11-07 [2] RSPM (R 4.4.0)
#> limma 3.62.1 2024-11-03 [1] Bioconductor 3.20 (R 4.4.2)
#> locfit * 1.5-9.10 2024-06-24 [1] RSPM (R 4.4.0)
#> lubridate 1.9.4 2024-12-08 [1] RSPM (R 4.4.0)
#> magrittr 2.0.3 2022-03-30 [2] RSPM (R 4.4.0)
#> Matrix 1.7-1 2024-10-18 [3] CRAN (R 4.4.2)
#> MatrixGenerics 1.18.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> matrixStats 1.4.1 2024-09-08 [1] RSPM (R 4.4.0)
#> memoise 2.0.1 2021-11-26 [2] RSPM (R 4.4.0)
#> mime 0.12 2021-09-28 [2] RSPM (R 4.4.0)
#> munsell 0.5.1 2024-04-01 [1] RSPM (R 4.4.0)
#> nnet 7.3-19 2023-05-03 [3] CRAN (R 4.4.2)
#> org.Hs.eg.db * 3.20.0 2024-12-12 [1] Bioconductor
#> OrganismDbi 1.48.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> pillar 1.9.0 2023-03-22 [2] RSPM (R 4.4.0)
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.4.0)
#> pkgdown 2.1.1 2024-09-17 [2] RSPM (R 4.4.0)
#> plyr 1.8.9 2023-10-02 [1] RSPM (R 4.4.0)
#> png 0.1-8 2022-11-29 [1] RSPM (R 4.4.0)
#> prettyunits 1.2.0 2023-09-24 [2] RSPM (R 4.4.0)
#> progress 1.2.3 2023-12-06 [1] RSPM (R 4.4.0)
#> ProtGenerics 1.38.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> purrr 1.0.2 2023-08-10 [2] RSPM (R 4.4.0)
#> qvalue 2.38.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> R6 2.5.1 2021-08-19 [2] RSPM (R 4.4.0)
#> ragg 1.3.3 2024-09-11 [2] RSPM (R 4.4.0)
#> rappdirs 0.3.3 2021-01-31 [2] RSPM (R 4.4.0)
#> RBGL 1.82.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> RColorBrewer 1.1-3 2022-04-03 [1] RSPM (R 4.4.0)
#> Rcpp 1.0.13-1 2024-11-02 [2] RSPM (R 4.4.0)
#> RCurl 1.98-1.16 2024-07-11 [1] RSPM (R 4.4.0)
#> RefManageR * 1.4.0 2022-09-30 [1] RSPM (R 4.4.0)
#> reshape2 1.4.4 2020-04-09 [1] RSPM (R 4.4.0)
#> restfulr 0.0.15 2022-06-16 [1] RSPM (R 4.4.0)
#> rjson 0.2.23 2024-09-16 [1] RSPM (R 4.4.0)
#> rlang 1.1.4 2024-06-04 [2] RSPM (R 4.4.0)
#> rmarkdown 2.29 2024-11-04 [2] RSPM (R 4.4.0)
#> rngtools 1.5.2 2021-09-20 [1] RSPM (R 4.4.0)
#> rpart 4.1.23 2023-12-05 [3] CRAN (R 4.4.2)
#> Rsamtools 2.22.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> RSQLite 2.3.9 2024-12-03 [1] RSPM (R 4.4.0)
#> rstudioapi 0.17.1 2024-10-22 [2] RSPM (R 4.4.0)
#> rtracklayer 1.66.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> S4Arrays 1.6.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> S4Vectors * 0.44.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> sass 0.4.9 2024-03-15 [2] RSPM (R 4.4.0)
#> scales 1.3.0 2023-11-28 [1] RSPM (R 4.4.0)
#> sessioninfo * 1.2.2 2021-12-06 [2] RSPM (R 4.4.0)
#> SparseArray 1.6.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> statmod 1.5.0 2023-01-06 [1] RSPM (R 4.4.0)
#> stringi 1.8.4 2024-05-06 [2] RSPM (R 4.4.0)
#> stringr 1.5.1 2023-11-14 [2] RSPM (R 4.4.0)
#> SummarizedExperiment 1.36.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> systemfonts 1.1.0 2024-05-15 [2] RSPM (R 4.4.0)
#> textshaping 0.4.1 2024-12-06 [2] RSPM (R 4.4.0)
#> tibble 3.2.1 2023-03-20 [2] RSPM (R 4.4.0)
#> tidyr 1.3.1 2024-01-24 [1] RSPM (R 4.4.0)
#> tidyselect 1.2.1 2024-03-11 [1] RSPM (R 4.4.0)
#> timechange 0.3.0 2024-01-18 [1] RSPM (R 4.4.0)
#> txdbmaker 1.2.1 2024-11-25 [1] Bioconductor 3.20 (R 4.4.2)
#> UCSC.utils 1.2.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> utf8 1.2.4 2023-10-22 [2] RSPM (R 4.4.0)
#> VariantAnnotation 1.52.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> vctrs 0.6.5 2023-12-01 [2] RSPM (R 4.4.0)
#> withr 3.0.2 2024-10-28 [2] RSPM (R 4.4.0)
#> xfun 0.49 2024-10-31 [2] RSPM (R 4.4.0)
#> XML 3.99-0.17 2024-06-25 [1] RSPM (R 4.4.0)
#> xml2 1.3.6 2023-12-04 [2] RSPM (R 4.4.0)
#> XVector 0.46.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#> yaml 2.3.10 2024-07-26 [2] RSPM (R 4.4.0)
#> zlibbioc 1.52.0 2024-10-29 [1] Bioconductor 3.20 (R 4.4.2)
#>
#> [1] /__w/_temp/Library
#> [2] /usr/local/lib/R/site-library
#> [3] /usr/local/lib/R/library
#>
#> ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────Bibliography
This vignette was generated using BiocStyle (Oleś, 2024), knitr (Xie, 2014) and rmarkdown (Allaire, Xie, Dervieux et al., 2024) running behind the scenes.
Citations made with RefManageR (McLean, 2017).
[1] J. Allaire, Y. Xie, C. Dervieux, et al. rmarkdown: Dynamic Documents for R. R package version 2.29. 2024. URL: https://github.com/rstudio/rmarkdown.
[2] S. Arora, M. Morgan, M. Carlson, et al. GenomeInfoDb: Utilities for manipulating chromosome and other ‘seqname’ identifiers. 2017. DOI: 10.18129/B9.bioc.GenomeInfoDb.
[3] Bioconductor Package Maintainer. AnnotationHubData: Transform public data resources into Bioconductor Data Structures. R package version 1.36.0. 2024. DOI: 10.18129/B9.bioc.AnnotationHubData. URL: https://bioconductor.org/packages/AnnotationHubData.
[4] M. Carlson. org.Hs.eg.db: Genome wide annotation for Human. R package version 3.20.0. 2024.
[5] L. Collado-Torres. Build and access GenomicState objects for use with derfinder tools from sources like Gencode. https://github.com/LieberInstitute/GenomicState - R package version 0.99.16. 2024. DOI: 10.18129/B9.bioc.GenomicState. URL: http://www.bioconductor.org/packages/GenomicState.
[6] L. Collado-Torres, A. Nellore, A. C. Frazee, et al. “Flexible expressed region analysis for RNA-seq with derfinder”. In: Nucl. Acids Res. (2017). DOI: 10.1093/nar/gkw852. URL: http://nar.oxfordjournals.org/content/early/2016/09/29/nar.gkw852.
[7] J. Hester and J. Bryan. glue: Interpreted String Literals. R package version 1.8.0, https://github.com/tidyverse/glue. 2024. URL: https://glue.tidyverse.org/.
[8] A. E. Jaffe, P. Murakami, H. Lee, et al. “Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies”. In: International journal of epidemiology 41.1 (2012), pp. 200–209. DOI: 10.1093/ije/dyr238.
[9] M. Lawrence, R. Gentleman, and V. Carey. “rtracklayer: an R package for interfacing with genome browsers”. In: Bioinformatics 25 (2009), pp. 1841-1842. DOI: 10.1093/bioinformatics/btp328. URL: http://bioinformatics.oxfordjournals.org/content/25/14/1841.abstract.
[10] M. Lawrence, W. Huber, H. Pagès, et al. “Software for Computing and Annotating Genomic Ranges”. In: PLoS Computational Biology 9 (8 2013). DOI: 10.1371/journal.pcbi.1003118. URL: http://www.ploscompbiol.org/article/info%3Adoi%2F10.1371%2Fjournal.pcbi.1003118}.
[11] M. W. McLean. “RefManageR: Import and Manage BibTeX and BibLaTeX References in R”. In: The Journal of Open Source Software (2017). DOI: 10.21105/joss.00338.
[12] M. Morgan and L. Shepherd. AnnotationHub: Client to access AnnotationHub resources. R package version 3.14.0. 2024. DOI: 10.18129/B9.bioc.AnnotationHub. URL: https://bioconductor.org/packages/AnnotationHub.
[13] A. Oleś. BiocStyle: Standard styles for vignettes and other Bioconductor documents. R package version 2.34.0. 2024. DOI: 10.18129/B9.bioc.BiocStyle. URL: https://bioconductor.org/packages/BiocStyle.
[14] H. Pagès, M. Carlson, P. Aboyoun, et al. txdbmaker: Tools for making TxDb objects from genomic annotations. R package version 1.2.1. 2024. DOI: 10.18129/B9.bioc.txdbmaker. URL: https://bioconductor.org/packages/txdbmaker.
[15] H. Pagès, M. Carlson, S. Falcon, et al. AnnotationDbi: Manipulation of SQLite-based annotations in Bioconductor. R package version 1.68.0. 2024. DOI: 10.18129/B9.bioc.AnnotationDbi. URL: https://bioconductor.org/packages/AnnotationDbi.
[16] R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Vienna, Austria, 2024. URL: https://www.R-project.org/.
[17] H. Wickham. “testthat: Get Started with Testing”. In: The R Journal 3 (2011), pp. 5–10. URL: https://journal.r-project.org/archive/2011-1/RJournal_2011-1_Wickham.pdf.
[18] H. Wickham, W. Chang, R. Flight, et al. sessioninfo: R Session Information. R package version 1.2.2, https://r-lib.github.io/sessioninfo/. 2021. URL: https://github.com/r-lib/sessioninfo#readme.
[19] Y. Xie. “knitr: A Comprehensive Tool for Reproducible Research in R”. In: Implementing Reproducible Computational Research. Ed. by V. Stodden, F. Leisch and R. D. Peng. ISBN 978-1466561595. Chapman and Hall/CRC, 2014.