How to find Total RNA Expression Genes (TREGs)
Louise A. Huuki-Myers
Lieber Institute for Brain Development, Johns Hopkins Medical Campuslahuuki@gmail.com
Leonardo Collado-Torres
Lieber Institute for Brain Development, Johns Hopkins Medical Campuslcolladotor@gmail.com
31 March 2026
Source:vignettes/finding_Total_RNA_Expression_Genes.Rmd
finding_Total_RNA_Expression_Genes.RmdNote: TREG is pronounced as a single word and fully capitalized, unlike Regulatory T cells, which are known as “Tregs” (pronounced “T-regs”). The work described here is unrelated to regulatory T cells.
Basics
Install TREG
R is an open-source statistical environment which can be
easily modified to enhance its functionality via packages. TREG is a
R package available via Bioconductor. R can be
installed on any operating system from CRAN after which you can install
TREG
by using the following commands in your R session:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("TREG")
## Check that you have a valid Bioconductor installation
BiocManager::valid()Required knowledge
TREG (Huuki-Myers and Collado-Torres, 2026) is based on many other packages and in particular in those that have implemented the infrastructure needed for dealing with single cell RNA sequencing data, visualization functions, and interactive data exploration. That is, packages like SummarizedExperiment that allow you to store the data.
If you are asking yourself the question “Where do I start using Bioconductor?” you might be interested in this blog post.
Asking for help
As package developers, we try to explain clearly how to use our
packages and in which order to use the functions. But R and
Bioconductor have a steep learning curve so it is critical
to learn where to ask for help. The blog post quoted above mentions some
but we would like to highlight the Bioconductor support site
as the main resource for getting help regarding Bioconductor. Other
alternatives are available such as creating GitHub issues and tweeting.
However, please note that if you want to receive help you should adhere
to the posting
guidelines. It is particularly critical that you provide a small
reproducible example and your session information so package developers
can track down the source of the error.
Citing TREG
We hope that TREG will be useful for your research. Please use the following information to cite the package and the research article describing the data provided by TREG. Thank you!
## Citation info
citation("TREG")
#> To cite package 'TREG' in publications use:
#>
#> Huuki-Myers LA, Collado-Torres L (2026). _TREG: a R/Bioconductor
#> package to identify Total RNA Expression Genes_.
#> doi:10.18129/B9.bioc.TREG <https://doi.org/10.18129/B9.bioc.TREG>.
#> https://github.com/LieberInstitute/TREG/TREG - R package version
#> 1.15.0, <http://www.bioconductor.org/packages/TREG>.
#>
#> Huuki-Myers LA, Montgomery KD, Kwon SH, Page SC, Hicks SC, Maynard
#> KR, Collado-Torres L (2022). "Data Driven Identification of Total RNA
#> Expression Genes "TREGs" for estimation of RNA abundance in
#> heterogeneous cell types." _bioRxiv_. doi:10.1101/2022.04.28.489923
#> <https://doi.org/10.1101/2022.04.28.489923>.
#> <https://doi.org/10.1101/2022.04.28.489923>.
#>
#> To see these entries in BibTeX format, use 'print(<citation>,
#> bibtex=TRUE)', 'toBibtex(.)', or set
#> 'options(citation.bibtex.max=999)'.Overview
The TREG (Huuki-Myers and Collado-Torres, 2026) package was developed for identifying candidate Total RNA Expression Genes (TREGs) for estimating RNA abundance for individual cells in an snFISH experiment by researchers at the Lieber Institute for Brain Development (LIBD) (Huuki-Myers, Montgomery, Kwon, Page, Hicks, Maynard, and Collado-Torres, 2022).
In this vignette we’ll showcase how you can use the R functions provided by TREG (Huuki-Myers and Collado-Torres, 2026) with the snRNA-seq dataset that was recently published by our LIBD collaborators (Tran, Maynard, Spangler, Huuki, Montgomery, Sadashivaiah, Tippani, Barry, Hancock, Hicks, Kleinman, Hyde, Collado-Torres, Jaffe, and Martinowich, 2021).
To get started, please load the TREG package.
The goal of TREG is to help find candidate Total
RNA Expression Genes (TREGs) in single nucleus (or single cell)
RNA-seq data.
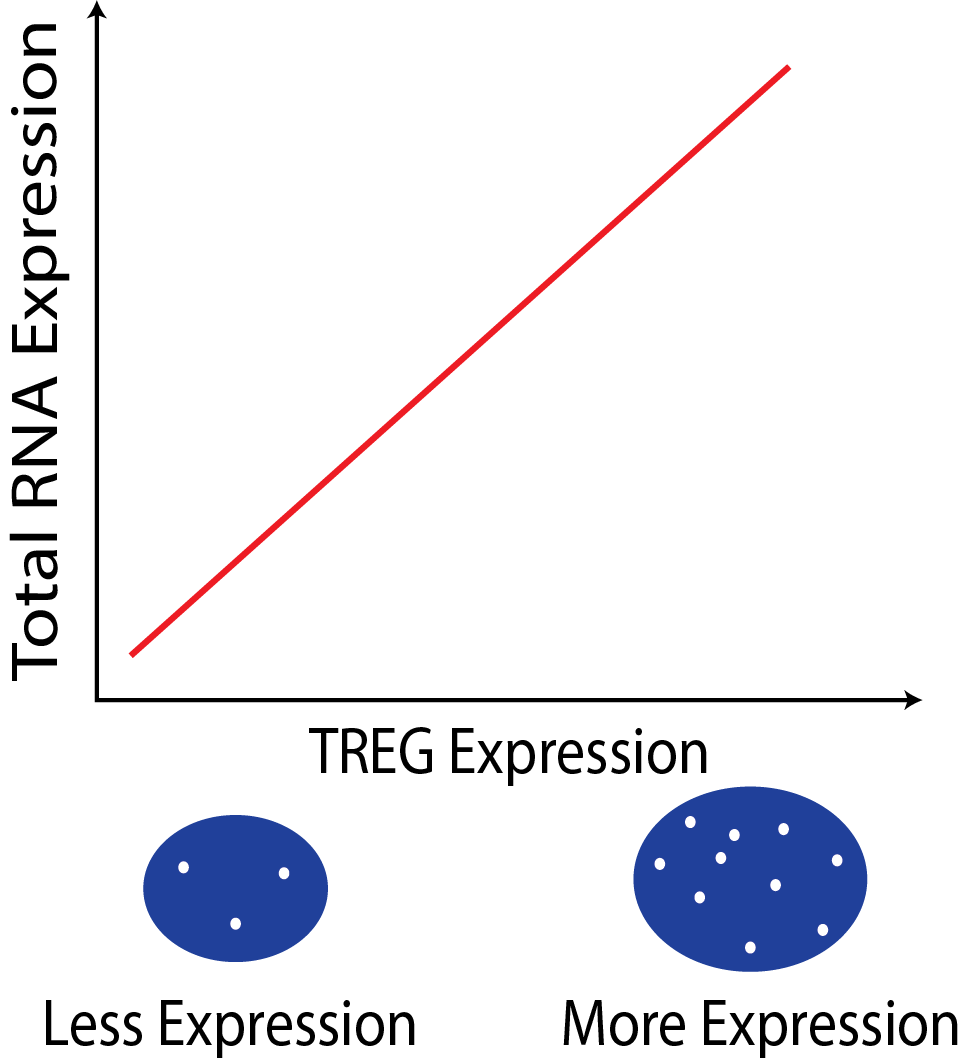
Why are TREGs useful?
The expression of a TREG is proportional to the the overall RNA expression in a cell. This relationship can be used to estimate total RNA content in cells in assays where only a few genes can be measured, such as single-molecule fluorescent in situ hybridization (smFISH).
In a smFISH experiment the number of TREG puncta can be used to infer the total RNA expression of the cell.
The motivation of this work is to collect data via smFISH in to help build better deconvolution algorithms. But may be many other application for TREGs in experimental design!
What makes a gene a good TREG?
The gene must have non-zero expression in most cells across different tissue and cell types.
A TREG should also be expressed at a constant level in respect to other genes across different cell types or have high rank invariance.
Be measurable as a continuous metric in the experimental assay, for example have a dynamic range of puncta when observed in RNAscope. This will need to be considered for the candidate TREGs, and may need to be validated experimentally.
How to find candidate TREGs with TREG
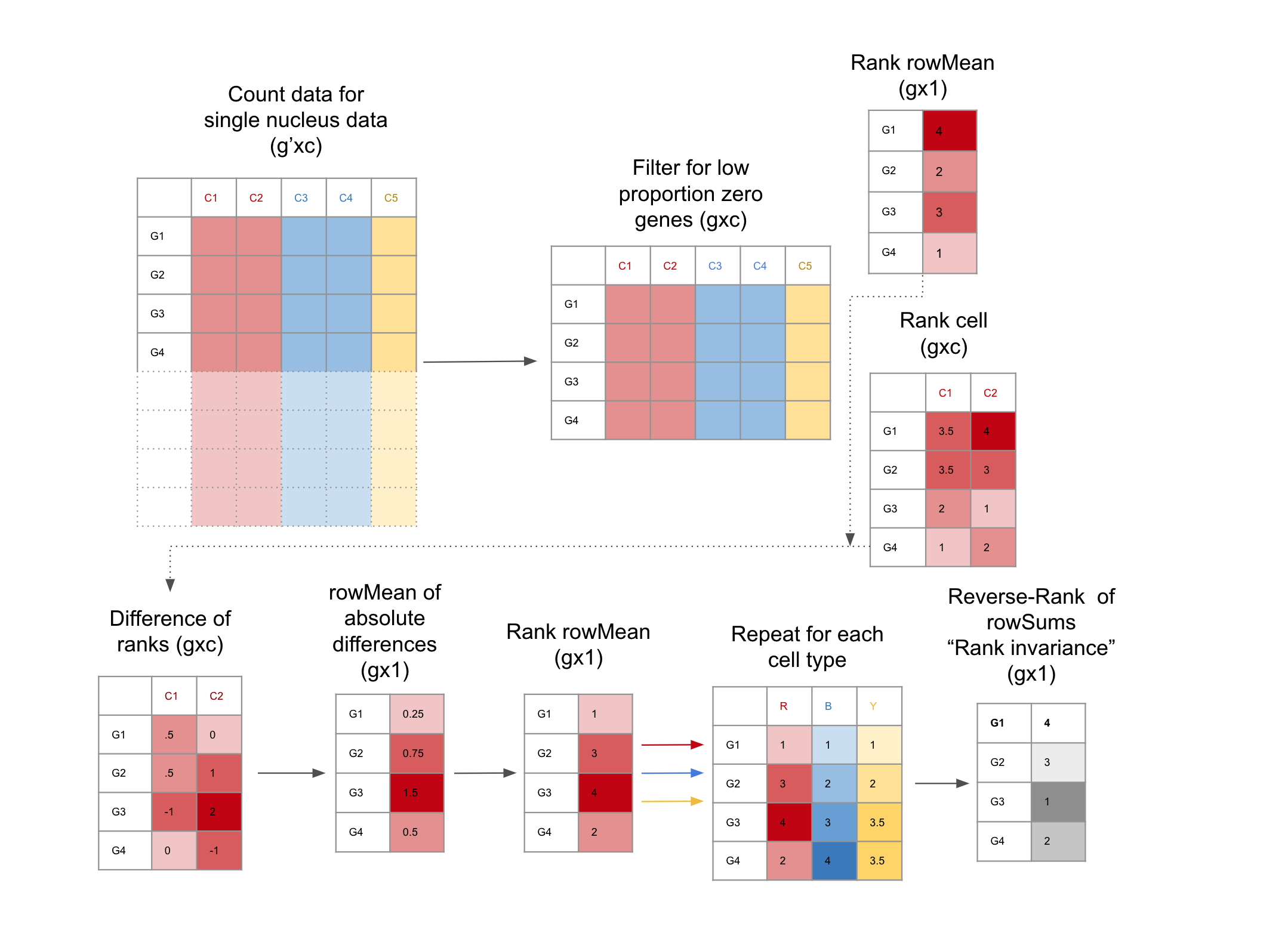
Filter for low Proportion Zero genes snRNA-seq dataset: This is facilitated with the functions
get_prop_zero()andfilter_prop_zero()(Equation @ref(eq:propZero)). snRNA-seq data is notoriously sparse, these functions enrich for genes with more universal expression.Evaluate genes for Rank Invariance The nuclei are grouped only by cell type. Within each cell type, the mean expression for each gene is ranked, the result is a vector (length is the number of genes), using the function
rank_group(). Then the expression of each gene is ranked for each nucleus,the result is a matrix (the number of nuclei x number of genes), using the functionrank_cells().Then the absolute difference between the rank of each nucleus and the mean expression is found, from here the mean of the differences for each gene is calculated, then ranked. These steps are repeated for each group, the result is a matrix of ranks, (number of cell types x number of genes). From here the sum of the ranks for each gene are reversed ranked, so there is one final value for each gene, the “Rank Invariance” The genes with the highest rank-invariance are considered good candidates as TREGs. This is calculated withrank_invariance_express(). This full process is implemented by:rank_invariance_express().
Example TREG Application
In this example we will apply our data driven process for TREG
discovery to a snRNA-seq dataset. This process has three main
steps:
1. Data prep
2. Gene filtering: dropping genes with low expression and high
Proportion Zero (Equation @ref(eq:propZero))
3. Rank Invariance Calculation
Download and Prep Data
Here we download a public single nucleus RNA-seq (snRNA-seq) data from (Tran, Maynard, Spangler et al., 2021) that we’ll use as our example. This data can be accessed on github. This data is from postmortem human brain in the dorsolateral prefrontal cortex (DLPFC) region, and contains gene expression data for 11k nuclei.
We will use BiocFileCache() to cache this data. It is
stored as a SingleCellExperiment object named
sce.dlpfc.tran, and takes 1.01 GB of RAM memory to
load.
# Download and save a local cache of the data available at:
# https://github.com/LieberInstitute/10xPilot_snRNAseq-human#processed-data
bfc <- BiocFileCache::BiocFileCache()
#> Registered S3 method overwritten by 'bit64':
#> method from
#> print.bitstring tools
url <- paste0(
"https://libd-snrnaseq-pilot.s3.us-east-2.amazonaws.com/",
"SCE_DLPFC-n3_tran-etal.rda"
)
local_data <- BiocFileCache::bfcrpath(url, x = bfc)
load(local_data, verbose = TRUE)
#> Loading objects:
#> sce.dlpfc.tran| Cell Type | Acronym |
|---|---|
| Astrocyte | Astro |
| Excitatory Neurons | Excit |
| Microglia | Micro |
| Oligodendrocytes | Oligo |
| Oligodendrocyte Progenitor Cells | OPC |
| Inhibitory Neurons | Inhib |
Human brain tissue consists of many types of cells, for the porpose of this demo, we will focus on the six major cell types listed in Table @ref(tab:acronyms).
Filter and Refine to Cell Types of Interest
First we will combine all of the Excit, Inhib subtypes, as it is a
finer resolution than we want to examine, and combine rare subtypes in
to one group. If there are too few cells in a group there may not be
enough data to get good results. This new cell type classification is
stored in the colData as cellType.broad.
## Explore the dimensions and cell type annotations
dim(sce.dlpfc.tran)
#> [1] 33538 11202
table(sce.dlpfc.tran$cellType)
#>
#> Astro Excit_A Excit_B Excit_C Excit_D Excit_E Excit_F
#> 782 529 773 524 132 187 243
#> Inhib_A Inhib_B Inhib_C Inhib_D Inhib_E Inhib_F Macrophage
#> 333 454 365 413 7 8 10
#> Micro Mural Oligo OPC Tcell
#> 388 18 5455 572 9
## Use a lower resolution of cell type annotation
sce.dlpfc.tran$cellType.broad <- gsub("_[A-Z]$", "", sce.dlpfc.tran$cellType)
(cell_type_tab <- table(sce.dlpfc.tran$cellType.broad))
#>
#> Astro Excit Inhib Macrophage Micro Mural Oligo
#> 782 2388 1580 10 388 18 5455
#> OPC Tcell
#> 572 9Next, we will drop any groups with < 50 cells after merging subtypes. This excludes any very rare cell types. Now we are working with the six broad cell types we are interested in.
## Find cell types with < 50 cells
(ct_drop <- names(cell_type_tab)[cell_type_tab < 50])
#> [1] "Macrophage" "Mural" "Tcell"
## Filter columns of sce object
sce.dlpfc.tran <- sce.dlpfc.tran[, !sce.dlpfc.tran$cellType.broad %in% ct_drop]
## Check new cell type bread down and dimension
table(sce.dlpfc.tran$cellType.broad)
#>
#> Astro Excit Inhib Micro Oligo OPC
#> 782 2388 1580 388 5455 572
dim(sce.dlpfc.tran)
#> [1] 33538 11165Filter Genes
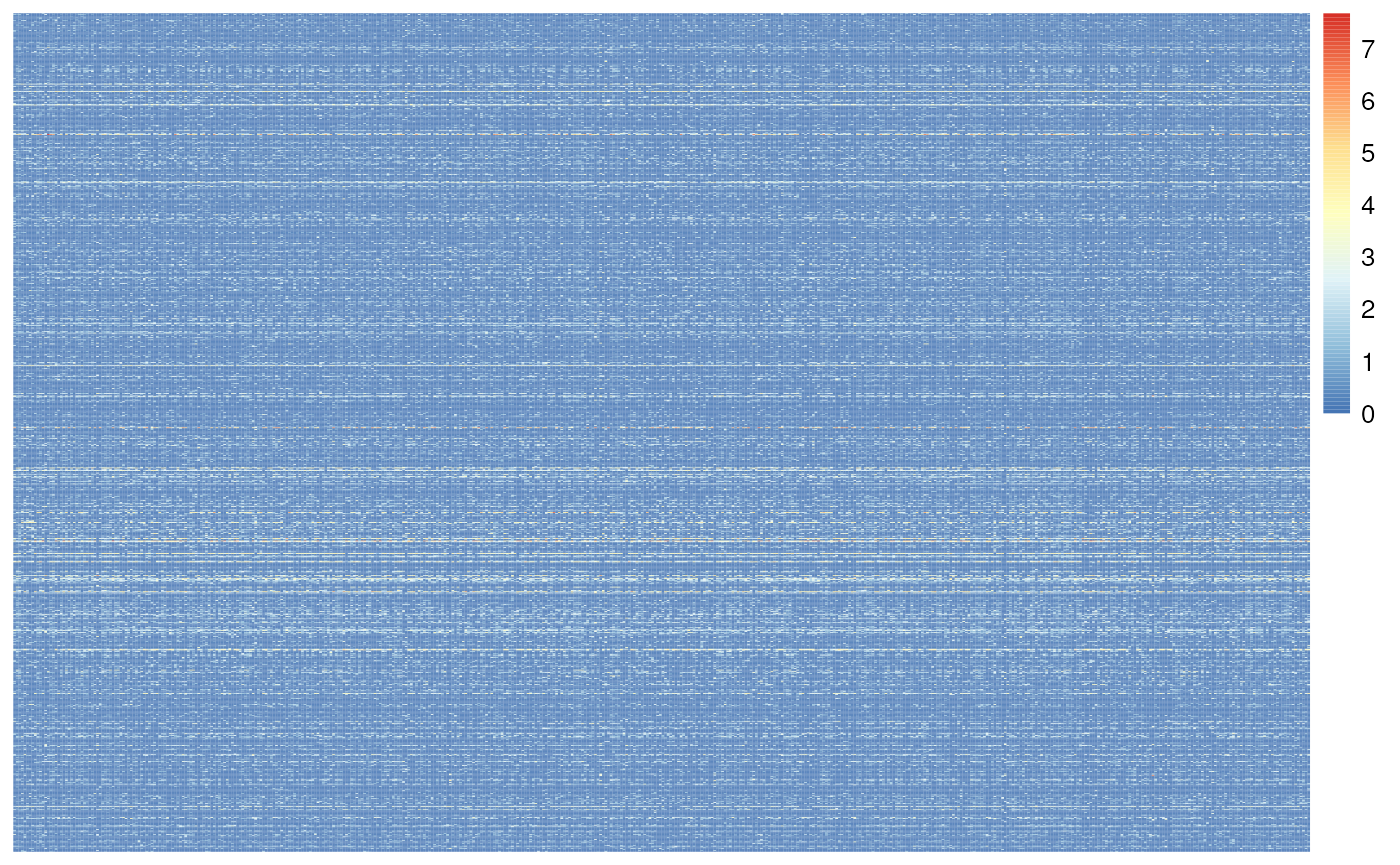
Single Nucleus data is often very sparse (lots of zeros in the count data), this dataset is 88% sparse. We can illustrate this in the heat map of the first 1k genes and 500 cells. The heatmap is mostly blue, indicating low values (Figure @ref(fig:examineSparsity)).
## this data is 88% sparse
sum(assays(sce.dlpfc.tran)$counts == 0) / (nrow(sce.dlpfc.tran) * ncol(sce.dlpfc.tran))
#> [1] 0.8839022
## lets make a heatmap of the first 1k genes and 500 cells
count_test <- as.matrix(assays(sce.dlpfc.tran)$logcounts[seq_len(1000), seq_len(500)])
pheatmap(count_test,
cluster_rows = FALSE,
cluster_cols = FALSE,
show_rownames = FALSE,
show_colnames = FALSE
)
Heatmap of the snRNA-seq counts. Illustrates sparseness of unfiltered data.
Filter to Top 50% Expression
Determine the median expression of genes over all rows, drop all the genes that are below this limit.
row_means <- rowMeans(assays(sce.dlpfc.tran)$logcounts)
(median_row_means <- median(row_means))
#> [1] 0.01405562
sce.dlpfc.tran <- sce.dlpfc.tran[row_means > median_row_means, ]
dim(sce.dlpfc.tran)
#> [1] 16769 11165After this filter lets check sparsity and make a heatmap of the first 1k genes and 500 cells. We are seeing more non-blue (Figure @ref(fig:top50FilterHeatmap))!
## this data down to 77% sparse
sum(assays(sce.dlpfc.tran)$counts == 0) / (nrow(sce.dlpfc.tran) * ncol(sce.dlpfc.tran))
#> [1] 0.7713423
## replot heatmap
count_test <- as.matrix(assays(sce.dlpfc.tran)$logcounts[seq_len(1000), seq_len(500)])
pheatmap::pheatmap(count_test,
cluster_rows = FALSE,
cluster_cols = FALSE,
show_rownames = FALSE,
show_colnames = FALSE
)
Heatmap of the snRNA-seq counts. With top 50% filtering the data becomes less sparse.
Calculate Proportion Zero and Pick Cutoff
For each group (let’s use cellType.broad) get the
Proportion Zero for each gene, where Proportion Zero is defined in
Equation @ref(eq:propZero) where
is the number of snRNA-seq counts for cell/nucleus
for gene
,
cell type
,
brain region
,
and
is the number of cells/nuclei for cell type
and brain region
.
# get prop zero for each gene for each cell type
prop_zeros <- get_prop_zero(sce.dlpfc.tran, group_col = "cellType.broad")
head(prop_zeros)
#> Astro Excit Inhib Micro Oligo OPC
#> AL627309.1 0.9667519 0.7784757 0.8487342 0.9536082 0.9552704 0.9685315
#> AL669831.5 0.8836317 0.2361809 0.4879747 0.8608247 0.8445463 0.7657343
#> LINC00115 0.9948849 0.9493300 0.9759494 0.9845361 0.9891842 0.9930070
#> NOC2L 0.9347826 0.6168342 0.7556962 0.9536082 0.9365720 0.8811189
#> KLHL17 0.9859335 0.8986600 0.9360759 0.9922680 0.9963336 0.9877622
#> HES4 0.8695652 0.7357621 0.6892405 0.9974227 0.9906508 0.9737762To determine a good cutoff for filtering lets examine the distribution of these Proportion Zeros by group.
# Pivot data longer for plotting
prop_zero_long <- prop_zeros %>%
rownames_to_column("Gene") %>%
pivot_longer(!Gene, names_to = "Group", values_to = "prop_zero")
# Plot histograms
(prop_zero_histogram <- ggplot(
data = prop_zero_long,
aes(x = prop_zero, fill = Group)
) +
geom_histogram(binwidth = 0.05) +
facet_wrap(~Group))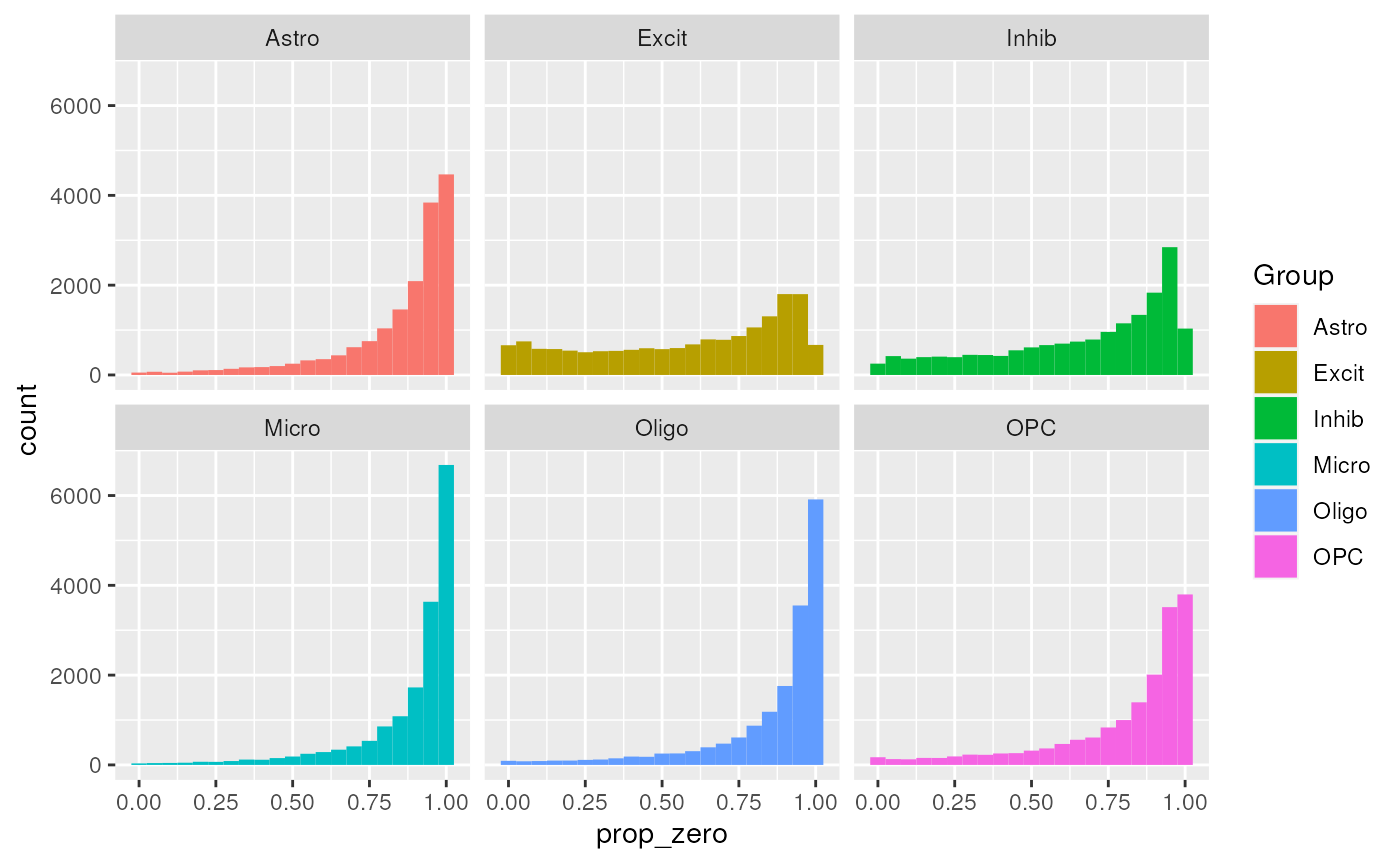
Distribution of Proption Zero across cell types and regions.
Looks like around 0.9 the densities peak, we’ll set that as the cutoff (Figure @ref(fig:propZeroDistribution)).
## Specify a cutoff, here we use 0.9
propZero_limit <- 0.9
## Add a vertical red dashed line where the cutoff is located
prop_zero_histogram +
geom_vline(xintercept = propZero_limit, color = "red", linetype = "dashed")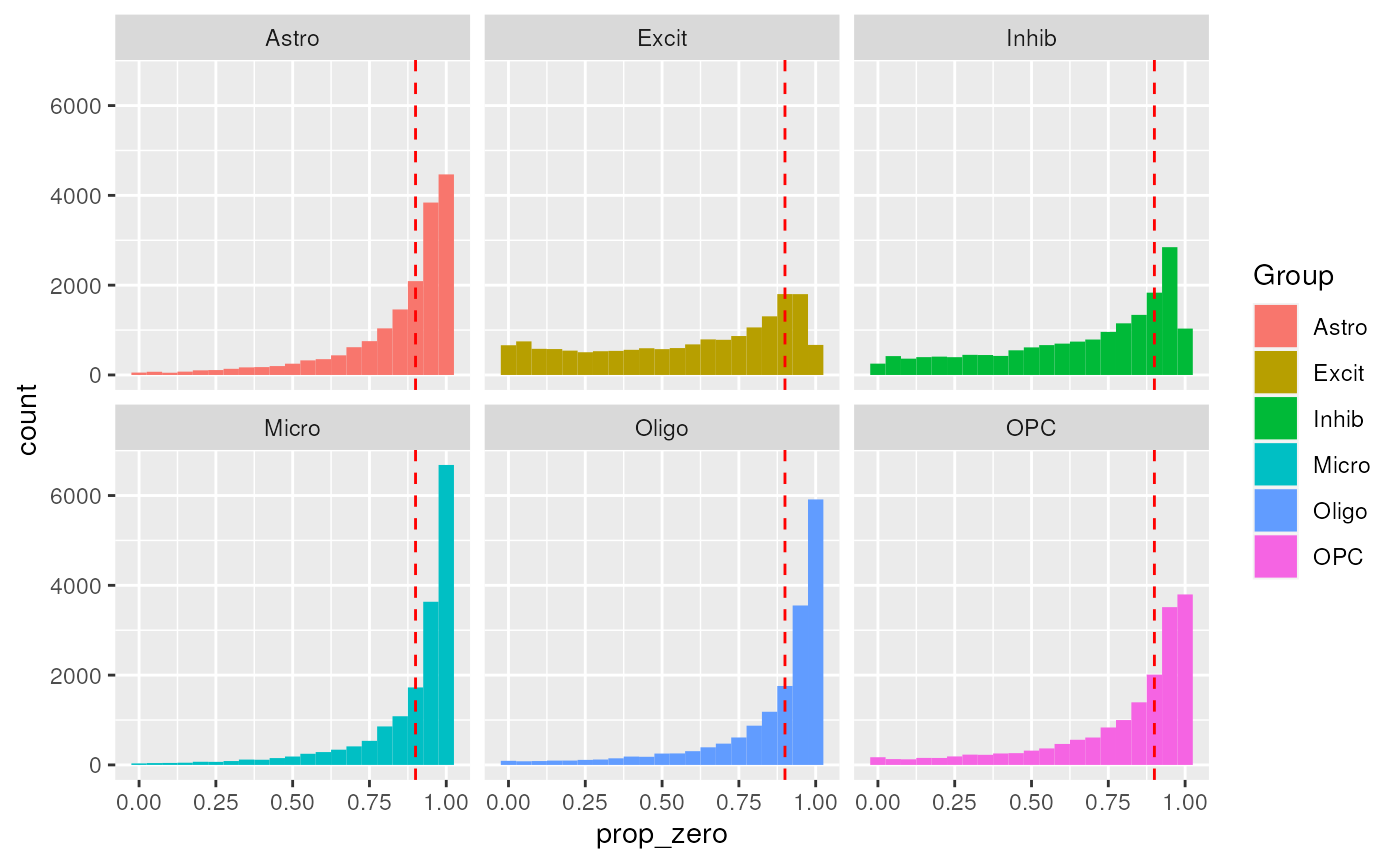
Show Proportion Zero Cutoff on Distributions.
The chosen cutoff excludes the peak Proportion Zeros from all groups (Figure @ref(fig:pickCutoff)).
Filter by the Max Proportion Zero
Use the cutoff to filter the remaining genes. Only 4k or ~11% of genes pass with this cutoff. Filter the SCE object to this set of genes.
## get a list of filtered genes
filtered_genes <- filter_prop_zero(prop_zeros, cutoff = propZero_limit)
## How many genes pass the filter?
length(filtered_genes)
#> [1] 4005
## What % of genes is this
length(filtered_genes) / nrow(sce.dlpfc.tran)
#> [1] 0.2388336
## Filter the sce object
sce.dlpfc.tran <- sce.dlpfc.tran[filtered_genes, ]One last check of the sparsity, more non-blue means more non-zero values for the Rank Invariance calculation, which prevents rank ties (Figure @ref(fig:propZeroFilterHeatmap)).
## this data down to 50% sparse
sum(assays(sce.dlpfc.tran)$counts == 0) / (nrow(sce.dlpfc.tran) * ncol(sce.dlpfc.tran))
#> [1] 0.5010587
## re-plot heatmap
count_test <- as.matrix(assays(sce.dlpfc.tran)$logcounts[seq_len(1000), seq_len(500)])
pheatmap::pheatmap(count_test,
cluster_rows = FALSE,
cluster_cols = FALSE,
show_rownames = FALSE,
show_colnames = FALSE
)
Heatmap of the snRNA-seq counts after Proption Zero filtering.
Run Rank Invariance
To get the Rank Invariance (RI), the rank of the genes across the cells in a group, and between groups is considered. One way to calculate RI is to find the group rank values, and cell rank values separately, then combine them as shown below. The genes with the top RI values are the best candidate TREGs.
## Get the rank of the gene in each group
group_rank <- rank_group(sce.dlpfc.tran, group_col = "cellType.broad")
## Get the rank of the gene for each cell
cell_rank <- rank_cells(sce.dlpfc.tran, group_col = "cellType.broad")
## Use both rankings to calculate rank_invariance()
rank_invar <- rank_invariance(group_rank, cell_rank)
## The top 5 Candidate TREGs:
head(sort(rank_invar, decreasing = TRUE))
#> MALAT1 CTNND2 FTX RERE FAM155A GNAQ
#> 4005 4004 4003 4002 4001 4000The rank_invariance_express() function combines these
three steps into one function, and achieves the same results.
## rank_invariance_express() runs the previous functions for you
rank_invar2 <- rank_invariance_express(
sce.dlpfc.tran,
group_col = "cellType.broad"
)
## Again the top 5 Candidate TREGs:
head(sort(rank_invar2, decreasing = TRUE))
#> MALAT1 CTNND2 FTX RERE FAM155A GNAQ
#> 4005 4004 4003 4002 4001 4000
## Check computationally that the results are identical
stopifnot(identical(rank_invar, rank_invar2))The rank_invariance_express() function is more efficient
as well, as it loops through the data to rank the genes over cells, and
groups at the same time.
Selecting thresholds
When identifying candidate TREGs with our software, there are a few thresholds users will select. In our manuscript (Huuki-Myers, Montgomery, Kwon et al., 2022), we used a few filters.
- We focused on genes among the top 50% of genes expressed in the snRNA-seq data. This helped address sparsity inherent to snRNA-seq data.
- We used a maximum Proportion Zero filter of 75%.
Overall it’s a balancing act between the computational requirements (reduce sparsity inherent to snRNA-seq data, reduce expression rank ties) and the biological goal (select a gene expressed in most nuclei from all cell types of interest). If you focus on just genes expressed in all nuclei or above a given threshold as shown in this figure, you could be losing too many genes (likely no gene is expressed in all nuclei as shown in our data) or genes that are not expressed at all in some cell types, given that some cell types are much less frequent than other cell types. While we consider the thresholds we used as those that balance both aspects and are practical, ultimately we do encourage users to plot their own data.
For example, making plots like those from Supplementary Figure 2. Supplementary Figure 2A is useful to examine whether genes you might have expected to pass the filters are being dropped. You can then check they were just below the filtering cutoffs, or significantly far away from them. Once you have the candidate TREG results, then Supplementary Figure 2B is useful to examine at what point do the top candidate TREGs have a stronger relationship with total RNA expression as measured from the snRNA-seq data. That is, where the blue curve jumps up and shows a more clear association between the two axes of that plot.
Among the top candidate TREGs, there might be practical limitations to consider for using that TREG in another assay, such as availability of RNAscope probes as well as measurability of the puncta for a given probe. We showed in Figure 5 how MALAT1 could not be reliably quantified with RNAscope due to high expression and oversaturation of fluorescent signals. In the case of RNAscope, we recommend testing the measurability of candidate TREGs with RNAscope data before generating a full dataset with a probe that may be difficult to accurately quantify.
Conclusion
We have identified top candidate TREG genes from this dataset, by
applying Proportion Zero filtering and calculating the Rank Invariance
using the TREG package. This provides a list of candidate
genes that can be useful for estimating total RNA expression in assays
such as smFISH.
However, we are unable to assess other important qualities of these genes that ensure they are experimentally compatible with the chosen assay. For example, in smFISH with RNAscope it is important that a TREG be expressed at a level that individual puncta can be accurately counted, and have a dynamic range of puncta. During experimental validation we found that MALAT1 was too highly expressed in the human DLPFC to segment individual puncta, and ruled it out as an experimentally useful TREG (Huuki-Myers, Montgomery, Kwon et al., 2022).
Therefore, we recommend that TREGs be evaluated in the assay or analysis of choice you perform a validation experiment with a pilot sample before implementing experiments using it on rare and valuable samples.
If you are designing a sc/snRNA-seq study to use as a reference for deconvolution of bulk RNA-seq, we recommend that you generate spatially-adjacent dissections in order to use them for RNAscope experiments. By doing so, you could identify cell types in your sc/snRNA-seq data, then identify candidate TREGs based on those cell types, and use these candidate TREGs in your spatially-adjacent dissections to quantify size and total RNA amounts for the cell types of interest (Huuki-Myers, Montgomery, Kwon et al., 2022). Furthermore, the RNAscope data alone can be used as a gold standard reference for cell fractions.
TREGs could be useful for other research purposes and other contexts than the ones we envisioned!
Thanks for your interest in TREGs :)
Reproducibility
The TREG package (Huuki-Myers and Collado-Torres, 2026) was made possible thanks to:
- R (R Core Team, 2026)
- BiocFileCache (Shepherd and Morgan, 2025)
- BiocStyle (Oleś, 2025)
- dplyr (Wickham, François, Henry, Müller, and Vaughan, 2026)
- ggplot2 (Wickham, 2016)
- knitr (Xie, 2025)
- Matrix (Bates, Maechler, and Jagan, 2026)
- pheatmap (Kolde, 2025)
- purrr (Wickham and Henry, 2026)
- rafalib (Irizarry and Love, 2025)
- RefManageR (McLean, 2017)
- rmarkdown (Allaire, Xie, Dervieux, McPherson, Luraschi, Ushey, Atkins, Wickham, Cheng, Chang, and Iannone, 2026)
- sessioninfo (Wickham, Chang, Flight, Müller, and Hester, 2025)
- SummarizedExperiment (Morgan, Obenchain, Hester, and Pagès, 2026)
- testthat (Wickham, 2011)
- tibble (Müller and Wickham, 2026)
- tidyr (Wickham, Vaughan, and Girlich, 2025)
Code for creating the vignette
## Create the vignette
library("rmarkdown")
system.time(render("finding_Total_RNA_Expression_Genes.Rmd"))
## Extract the R code
library("knitr")
knit("finding_Total_RNA_Expression_Genes.Rmd", tangle = TRUE)Date the vignette was generated.
#> [1] "2026-03-31 18:01:05 UTC"Wallclock time spent generating the vignette.
#> Time difference of 50.717 secsR session information.
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
#> version R Under development (unstable) (2026-03-28 r89738)
#> os Ubuntu 24.04.4 LTS
#> system x86_64, linux-gnu
#> ui X11
#> language en
#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
#> date 2026-03-31
#> pandoc 3.9.0.2 @ /usr/bin/ (via rmarkdown)
#> quarto 1.8.25 @ /usr/local/bin/quarto
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
#> abind 1.4-8 2024-09-12 [1] CRAN (R 4.6.0)
#> backports 1.5.0 2024-05-23 [1] CRAN (R 4.6.0)
#> bibtex 0.5.2 2026-02-03 [1] CRAN (R 4.6.0)
#> Biobase * 2.71.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
#> BiocFileCache 3.1.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
#> BiocGenerics * 0.57.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
#> BiocManager 1.30.27 2025-11-14 [2] CRAN (R 4.7.0)
#> BiocStyle * 2.39.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
#> bit 4.6.0 2025-03-06 [1] CRAN (R 4.6.0)
#> bit64 4.6.0-1 2025-01-16 [1] CRAN (R 4.6.0)
#> blob 1.3.0 2026-01-14 [1] CRAN (R 4.6.0)
#> bookdown 0.46 2025-12-05 [1] CRAN (R 4.6.0)
#> bslib 0.10.0 2026-01-26 [2] CRAN (R 4.7.0)
#> cachem 1.1.0 2024-05-16 [2] CRAN (R 4.7.0)
#> cli 3.6.5 2025-04-23 [2] CRAN (R 4.7.0)
#> curl 7.0.0 2025-08-19 [2] CRAN (R 4.7.0)
#> DBI 1.3.0 2026-02-25 [1] CRAN (R 4.7.0)
#> dbplyr 2.5.2 2026-02-13 [1] CRAN (R 4.7.0)
#> DelayedArray 0.37.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
#> desc 1.4.3 2023-12-10 [2] CRAN (R 4.7.0)
#> digest 0.6.39 2025-11-19 [2] CRAN (R 4.7.0)
#> dplyr * 1.2.0 2026-02-03 [1] CRAN (R 4.6.0)
#> evaluate 1.0.5 2025-08-27 [2] CRAN (R 4.7.0)
#> farver 2.1.2 2024-05-13 [1] CRAN (R 4.6.0)
#> fastmap 1.2.0 2024-05-15 [2] CRAN (R 4.7.0)
#> filelock 1.0.3 2023-12-11 [1] CRAN (R 4.6.0)
#> fs 2.0.1 2026-03-24 [2] CRAN (R 4.7.0)
#> generics * 0.1.4 2025-05-09 [1] CRAN (R 4.6.0)
#> GenomicRanges * 1.63.1 2025-12-08 [1] Bioconductor 3.23 (R 4.6.0)
#> ggplot2 * 4.0.2 2026-02-03 [1] CRAN (R 4.6.0)
#> glue 1.8.0 2024-09-30 [2] CRAN (R 4.7.0)
#> gtable 0.3.6 2024-10-25 [1] CRAN (R 4.6.0)
#> htmltools 0.5.9 2025-12-04 [2] CRAN (R 4.7.0)
#> htmlwidgets 1.6.4 2023-12-06 [2] CRAN (R 4.7.0)
#> httr 1.4.8 2026-02-13 [1] CRAN (R 4.7.0)
#> httr2 1.2.2 2025-12-08 [2] CRAN (R 4.7.0)
#> IRanges * 2.45.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
#> jquerylib 0.1.4 2021-04-26 [2] CRAN (R 4.7.0)
#> jsonlite 2.0.0 2025-03-27 [2] CRAN (R 4.7.0)
#> knitr 1.51 2025-12-20 [2] CRAN (R 4.7.0)
#> labeling 0.4.3 2023-08-29 [1] CRAN (R 4.6.0)
#> lattice 0.22-9 2026-02-09 [3] CRAN (R 4.7.0)
#> lifecycle 1.0.5 2026-01-08 [2] CRAN (R 4.7.0)
#> lubridate 1.9.5 2026-02-04 [1] CRAN (R 4.6.0)
#> magrittr 2.0.4 2025-09-12 [2] CRAN (R 4.7.0)
#> Matrix 1.7-5 2026-03-21 [3] CRAN (R 4.7.0)
#> MatrixGenerics * 1.23.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
#> matrixStats * 1.5.0 2025-01-07 [1] CRAN (R 4.6.0)
#> memoise 2.0.1 2021-11-26 [2] CRAN (R 4.7.0)
#> otel 0.2.0 2025-08-29 [2] CRAN (R 4.7.0)
#> pheatmap * 1.0.13 2025-06-05 [1] CRAN (R 4.6.0)
#> pillar 1.11.1 2025-09-17 [2] CRAN (R 4.7.0)
#> pkgconfig 2.0.3 2019-09-22 [2] CRAN (R 4.7.0)
#> pkgdown 2.2.0.9000 2026-03-31 [1] Github (r-lib/pkgdown@a6abe43)
#> plyr 1.8.9 2023-10-02 [1] CRAN (R 4.6.0)
#> purrr 1.2.1 2026-01-09 [2] CRAN (R 4.7.0)
#> R6 2.6.1 2025-02-15 [2] CRAN (R 4.7.0)
#> rafalib 1.0.4 2025-04-08 [1] CRAN (R 4.6.0)
#> ragg 1.5.2 2026-03-23 [2] CRAN (R 4.7.0)
#> rappdirs 0.3.4 2026-01-17 [2] CRAN (R 4.7.0)
#> RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.6.0)
#> Rcpp 1.1.1 2026-01-10 [2] CRAN (R 4.7.0)
#> RefManageR * 1.4.0 2022-09-30 [1] CRAN (R 4.6.0)
#> rlang 1.1.7 2026-01-09 [2] CRAN (R 4.7.0)
#> rmarkdown 2.31 2026-03-26 [2] CRAN (R 4.7.0)
#> RSQLite 2.4.6 2026-02-06 [1] CRAN (R 4.6.0)
#> S4Arrays 1.11.1 2025-11-25 [1] Bioconductor 3.23 (R 4.6.0)
#> S4Vectors * 0.49.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
#> S7 0.2.1 2025-11-14 [1] CRAN (R 4.6.0)
#> sass 0.4.10 2025-04-11 [2] CRAN (R 4.7.0)
#> scales 1.4.0 2025-04-24 [1] CRAN (R 4.6.0)
#> Seqinfo * 1.1.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
#> sessioninfo * 1.2.3 2025-02-05 [2] CRAN (R 4.7.0)
#> SingleCellExperiment * 1.33.2 2026-03-24 [1] Bioconductor 3.23 (R 4.7.0)
#> SparseArray 1.11.12 2026-03-30 [1] Bioconductor 3.23 (R 4.7.0)
#> stringi 1.8.7 2025-03-27 [2] CRAN (R 4.7.0)
#> stringr 1.6.0 2025-11-04 [2] CRAN (R 4.7.0)
#> SummarizedExperiment * 1.41.1 2026-02-06 [1] Bioconductor 3.23 (R 4.6.0)
#> systemfonts 1.3.2 2026-03-05 [2] CRAN (R 4.7.0)
#> textshaping 1.0.5 2026-03-06 [2] CRAN (R 4.7.0)
#> tibble * 3.3.1 2026-01-11 [2] CRAN (R 4.7.0)
#> tidyr * 1.3.2 2025-12-19 [1] CRAN (R 4.6.0)
#> tidyselect 1.2.1 2024-03-11 [1] CRAN (R 4.6.0)
#> timechange 0.4.0 2026-01-29 [1] CRAN (R 4.6.0)
#> TREG * 1.15.0 2026-03-31 [1] Bioconductor
#> vctrs 0.7.2 2026-03-21 [2] CRAN (R 4.7.0)
#> withr 3.0.2 2024-10-28 [2] CRAN (R 4.7.0)
#> xfun 0.57 2026-03-20 [2] CRAN (R 4.7.0)
#> xml2 1.5.2 2026-01-17 [2] CRAN (R 4.7.0)
#> XVector 0.51.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
#> yaml 2.3.12 2025-12-10 [2] CRAN (R 4.7.0)
#>
#> [1] /__w/_temp/Library
#> [2] /usr/local/lib/R/site-library
#> [3] /usr/local/lib/R/library
#> * ── Packages attached to the search path.
#>
#> ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────Bibliography
This vignette was generated using BiocStyle (Oleś, 2025), knitr (Xie, 2025) and rmarkdown (Allaire, Xie, Dervieux et al., 2026) running behind the scenes.
Citations made with RefManageR (McLean, 2017).
[1] J. Allaire, Y. Xie, C. Dervieux, et al. rmarkdown: Dynamic Documents for R. R package version 2.31. 2026. URL: https://github.com/rstudio/rmarkdown.
[2] D. Bates, M. Maechler, and M. Jagan. Matrix: Sparse and Dense Matrix Classes and Methods. R package version 1.7-5. 2026. DOI: 10.32614/CRAN.package.Matrix. URL: https://CRAN.R-project.org/package=Matrix.
[3] L. A. Huuki-Myers and L. Collado-Torres. TREG: a R/Bioconductor package to identify Total RNA Expression Genes. https://github.com/LieberInstitute/TREG/TREG - R package version 1.15.0. 2026. DOI: 10.18129/B9.bioc.TREG. URL: http://www.bioconductor.org/packages/TREG.
[4] L. A. Huuki-Myers, K. D. Montgomery, S. H. Kwon, et al. “Data Driven Identification of Total RNA Expression Genes "TREGs" for estimation of RNA abundance in heterogeneous cell types”. In: bioRxiv (2022). DOI: 10.1101/2022.04.28.489923. URL: https://doi.org/10.1101/2022.04.28.489923.
[5] R. A. Irizarry and M. I. Love. rafalib: Convenience Functions for Routine Data Exploration. R package version 1.0.4. 2025. DOI: 10.32614/CRAN.package.rafalib. URL: https://CRAN.R-project.org/package=rafalib.
[6] R. Kolde. pheatmap: Pretty Heatmaps. R package version 1.0.13. 2025. DOI: 10.32614/CRAN.package.pheatmap. URL: https://CRAN.R-project.org/package=pheatmap.
[7] M. W. McLean. “RefManageR: Import and Manage BibTeX and BibLaTeX References in R”. In: The Journal of Open Source Software (2017). DOI: 10.21105/joss.00338.
[8] M. Morgan, V. Obenchain, J. Hester, et al. SummarizedExperiment: A container (S4 class) for matrix-like assays. R package version 1.41.1. 2026. DOI: 10.18129/B9.bioc.SummarizedExperiment. URL: https://bioconductor.org/packages/SummarizedExperiment.
[9] K. Müller and H. Wickham. tibble: Simple Data Frames. R package version 3.3.1. 2026. DOI: 10.32614/CRAN.package.tibble. URL: https://CRAN.R-project.org/package=tibble.
[10] A. Oleś. BiocStyle: Standard styles for vignettes and other Bioconductor documents. R package version 2.39.0. 2025. DOI: 10.18129/B9.bioc.BiocStyle. URL: https://bioconductor.org/packages/BiocStyle.
[11] R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing (ROR: <https://ror.org/05qewa988>;). Vienna, Austria, 2026. DOI: 10.32614/R.manuals. URL: https://www.R-project.org/.
[12] L. Shepherd and M. Morgan. BiocFileCache: Manage Files Across Sessions. R package version 3.1.0. 2025. DOI: 10.18129/B9.bioc.BiocFileCache. URL: https://bioconductor.org/packages/BiocFileCache.
[13] M. N. Tran, K. R. Maynard, A. Spangler, et al. “Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain”. In: Neuron (2021). DOI: 10.1016/j.neuron.2021.09.001.
[14] H. Wickham. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York, 2016. ISBN: 978-3-319-24277-4. URL: https://ggplot2.tidyverse.org.
[15] H. Wickham. “testthat: Get Started with Testing”. In: The R Journal 3 (2011), pp. 5–10. URL: https://journal.r-project.org/articles/RJ-2011-002/.
[16] H. Wickham, W. Chang, R. Flight, et al. sessioninfo: R Session Information. R package version 1.2.3. 2025. DOI: 10.32614/CRAN.package.sessioninfo. URL: https://CRAN.R-project.org/package=sessioninfo.
[17] H. Wickham, R. François, L. Henry, et al. dplyr: A Grammar of Data Manipulation. R package version 1.2.0. 2026. DOI: 10.32614/CRAN.package.dplyr. URL: https://CRAN.R-project.org/package=dplyr.
[18] H. Wickham and L. Henry. purrr: Functional Programming Tools. R package version 1.2.1. 2026. DOI: 10.32614/CRAN.package.purrr. URL: https://CRAN.R-project.org/package=purrr.
[19] H. Wickham, D. Vaughan, and M. Girlich. tidyr: Tidy Messy Data. R package version 1.3.2. 2025. DOI: 10.32614/CRAN.package.tidyr. URL: https://CRAN.R-project.org/package=tidyr.
[20] Y. Xie. knitr: A General-Purpose Package for Dynamic Report Generation in R. R package version 1.51. 2025. URL: https://yihui.org/knitr/.