The goal of TREG is to help find candidate Total RNA Expression Genes (TREGs) in single nucleus (or single cell) RNA-seq data.
Note: TREG is pronounced as a single word and fully capitalized, unlike Regulatory T cells, which are known as “Tregs” (pronounced “T-regs”). The work described here is unrelated to regulatory T cells.
Why are TREGs useful?
The expression of a TREG is proportional to the the overall RNA expression in a cell. This relationship can be used to estimate total RNA content in cells in assays where only a few genes can be measured, such as single-molecule fluorescent in situ hybridization (smFISH).
In a smFISH experiment the number of TREG puncta can be used to infer the total RNA expression of the cell.
The motivation of this work is to collect data via smFISH in to help build better deconvolution algorithms. But may be many other application for TREGs in experimental design!
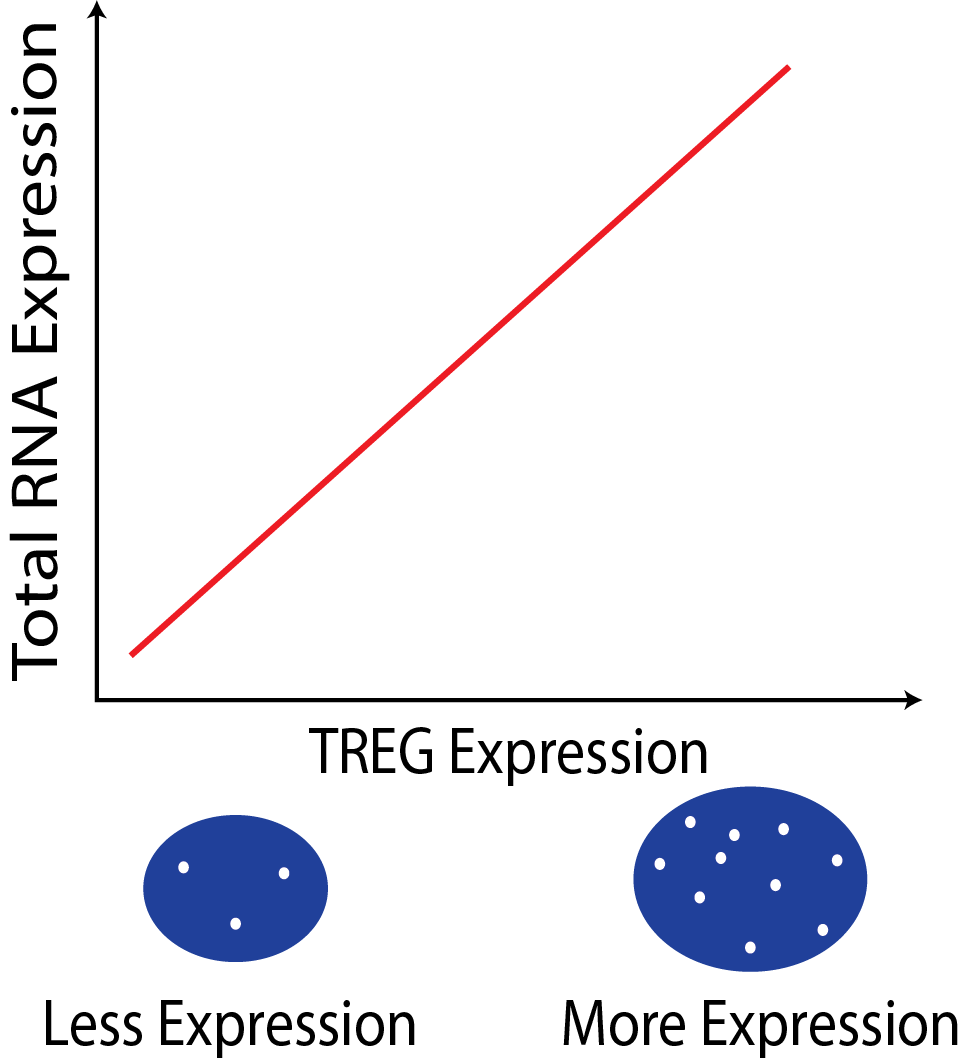
What makes a gene a good TREG?
The gene must have non-zero expression in most cells across different tissue and cell types.
A TREG should also be expressed at a constant level in respect to other genes across different cell types or have high rank invariance.
Be measurable as a continuous metric in the experimental assay, for example have a dynamic range of puncta when observed in RNAscope. This will need to be considered for the candidate TREGs, and may need to be validated experimentally.
![]()
How to find candidate TREGs with TREG
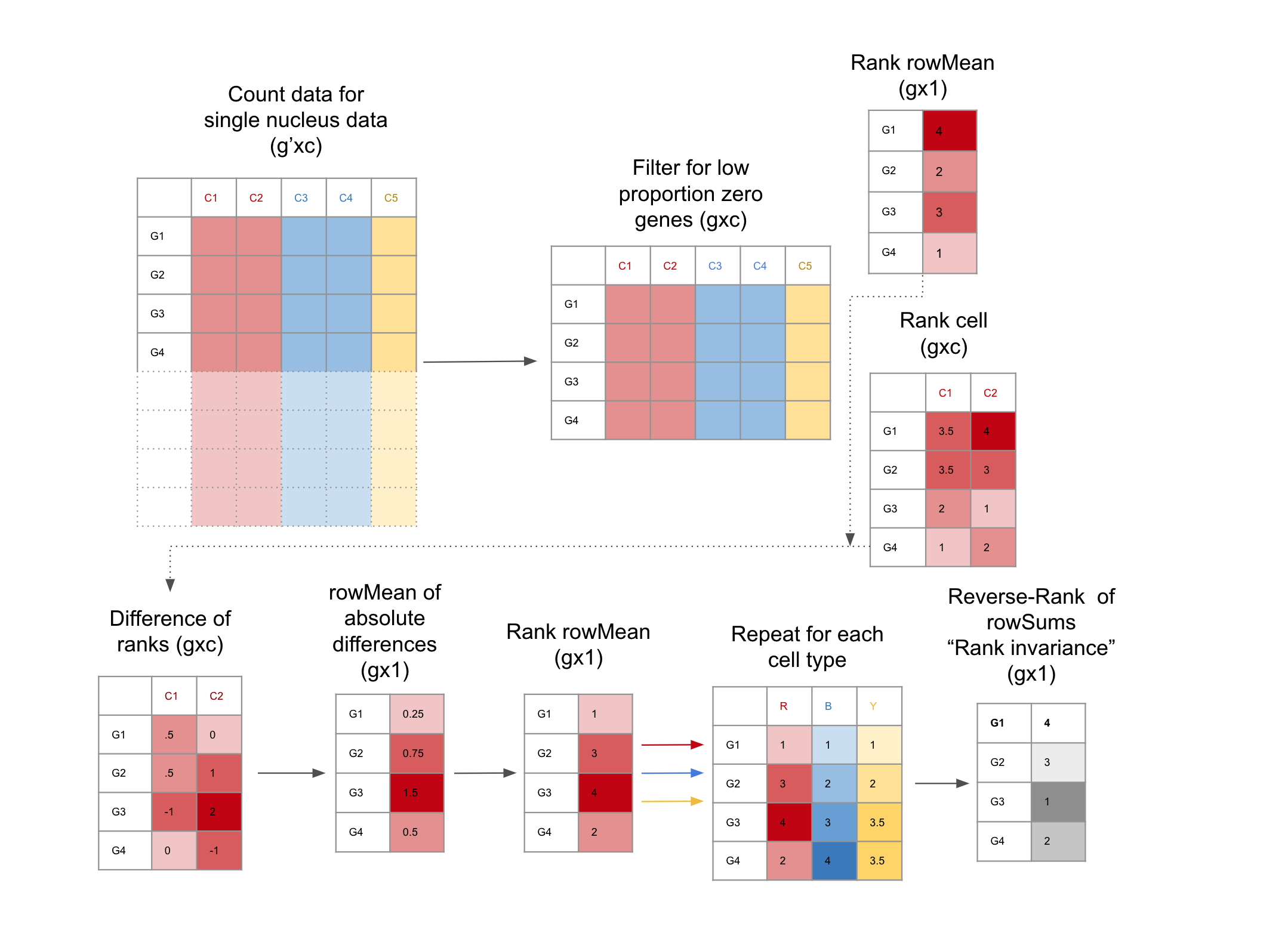
Filter for low Proportion Zero genes snRNA-seq dataset: This is facilitated with the functions
get_prop_zero()andfilter_prop_zero(). snRNA-seq data is notoriously sparse, these functions enrich for genes with more universal expression.Evaluate genes for Rank Invariance The nuclei are grouped only by cell type. Within each cell type, the mean expression for each gene is ranked, the result is a vector (length is the number of genes), using the function
rank_group(). Then the expression of each gene is ranked for each nucleus,the result is a matrix (the number of nuclei x number of genes), using the functionrank_cells().Then the absolute difference between the rank of each nucleus and the mean expression is found, from here the mean of the differences for each gene is calculated, then ranked. These steps are repeated for each group, the result is a matrix of ranks, (number of cell types x number of genes). From here the sum of the ranks for each gene are reversed ranked, so there is one final value for each gene, the “Rank Invariance” The genes with the highest rank-invariance are considered good candidates as TREGs. This is calculated withrank_invariance_express(). This full process is implemented by:rank_invariance_express().
Installation instructions
Get the latest stable R release from CRAN. Then install TREG using from Bioconductor the following code:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("TREG")And the development version from GitHub with:
BiocManager::install("LieberInstitute/TREG")Example
Proportion Zero Filtering
A TREG gene should be expressed in almost every cell. The set of genes should be filtered by maximum Proportion Zero within a groups of cells.
## Calculate Proportion Zero in groups defined by a column in colData
(prop_zero <- get_prop_zero(sce = sce_zero_test, group_col = "cellType"))
#> A B
#> g100 1.00 1.00
#> g50 0.48 0.52
#> g0 0.00 0.00
#> gOffOn 0.50 0.50
#> gVar 0.58 0.36
## Get list of genes that pass the max Proportion Zero filter
(filtered_genes <- filter_prop_zero(prop_zero, cutoff = 0.9))
#> [1] "g50" "g0" "gOffOn" "gVar"
## Filter sce object to this list of genes
sce_filter <- sce_zero_test[filtered_genes, ]Evaluate RI for Filtered SCE Data
The genes with the highest Rank Invariance are considered good candidates as TREGs. In this example the gene g0 would be the strongest candidate TREG.
## Get the Rank Invariance value for each gene
## The highest values are the best TREG candidates
ri <- rank_invariance_express(sce_filter)
sort(ri, decreasing = TRUE)
#> g0 gOffOn gVar g50
#> 4 3 2 1Citation
Below is the citation output from using citation('TREG') in R. Please run this yourself to check for any updates on how to cite TREG.
print(citation("TREG"), bibtex = TRUE)
#> To cite package 'TREG' in publications use:
#>
#> Huuki-Myers LA, Collado-Torres L (2023). _TREG: a R/Bioconductor
#> package to identify Total RNA Expression Genes_.
#> doi:10.18129/B9.bioc.TREG <https://doi.org/10.18129/B9.bioc.TREG>,
#> https://github.com/LieberInstitute/TREG/TREG - R package version
#> 1.5.1, <http://www.bioconductor.org/packages/TREG>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Manual{,
#> title = {TREG: a R/Bioconductor package to identify Total RNA Expression Genes},
#> author = {Louise A. Huuki-Myers and Leonardo Collado-Torres},
#> year = {2023},
#> url = {http://www.bioconductor.org/packages/TREG},
#> note = {https://github.com/LieberInstitute/TREG/TREG - R package version 1.5.1},
#> doi = {10.18129/B9.bioc.TREG},
#> }
#>
#> Huuki-Myers LA, Montgomery KD, Kwon SH, Page SC, Hicks SC, Maynard
#> KR, Collado-Torres L (2022). "Data Driven Identification of Total RNA
#> Expression Genes "TREGs" for estimation of RNA abundance in
#> heterogeneous cell types." _bioRxiv_. doi:10.1101/2022.04.28.489923
#> <https://doi.org/10.1101/2022.04.28.489923>,
#> <https://doi.org/10.1101/2022.04.28.489923>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Article{,
#> title = {Data Driven Identification of Total RNA Expression Genes "TREGs" for estimation of RNA abundance in heterogeneous cell types},
#> author = {Louise A. Huuki-Myers and Kelsey D. Montgomery and Sang Ho. Kwon and Stephanie C. Page and Stephanie C. Hicks and Kristen R. Maynard and Leonardo Collado-Torres},
#> year = {2022},
#> journal = {bioRxiv},
#> doi = {10.1101/2022.04.28.489923},
#> url = {https://doi.org/10.1101/2022.04.28.489923},
#> }Please note that the TREG was only made possible thanks to many other R and bioinformatics software authors, which are cited either in the vignettes and/or the paper(s) describing this package.
Code of Conduct
Please note that the TREG project is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.
Development tools
- Continuous code testing is possible thanks to GitHub actions through usethis, remotes, and rcmdcheck customized to use Bioconductor’s docker containers and BiocCheck.
- Code coverage assessment is possible thanks to codecov and covr.
- The documentation website is automatically updated thanks to pkgdown.
- The code is styled automatically thanks to styler.
- The documentation is formatted thanks to devtools and roxygen2.
For more details, check the dev directory.
This package was developed using biocthis.








