Introduction to Scraping and Wrangling Tables from Research Articles
By Steve Semick.
What do you do when you want to use results from the literature to anchor your own analysis? When these results are in the form of an easily accessible table, such as a .csv file or .xlsx file, then it is simple enough to download them and incorporate them into your research. Often times, however, published findings are not so easy to handle. Today, we’ll go through a practical scenario on scraping an html table from a Nature Genetics article into R and wrangling the data into a useful format. This is what the online table we want to scrape looks like:
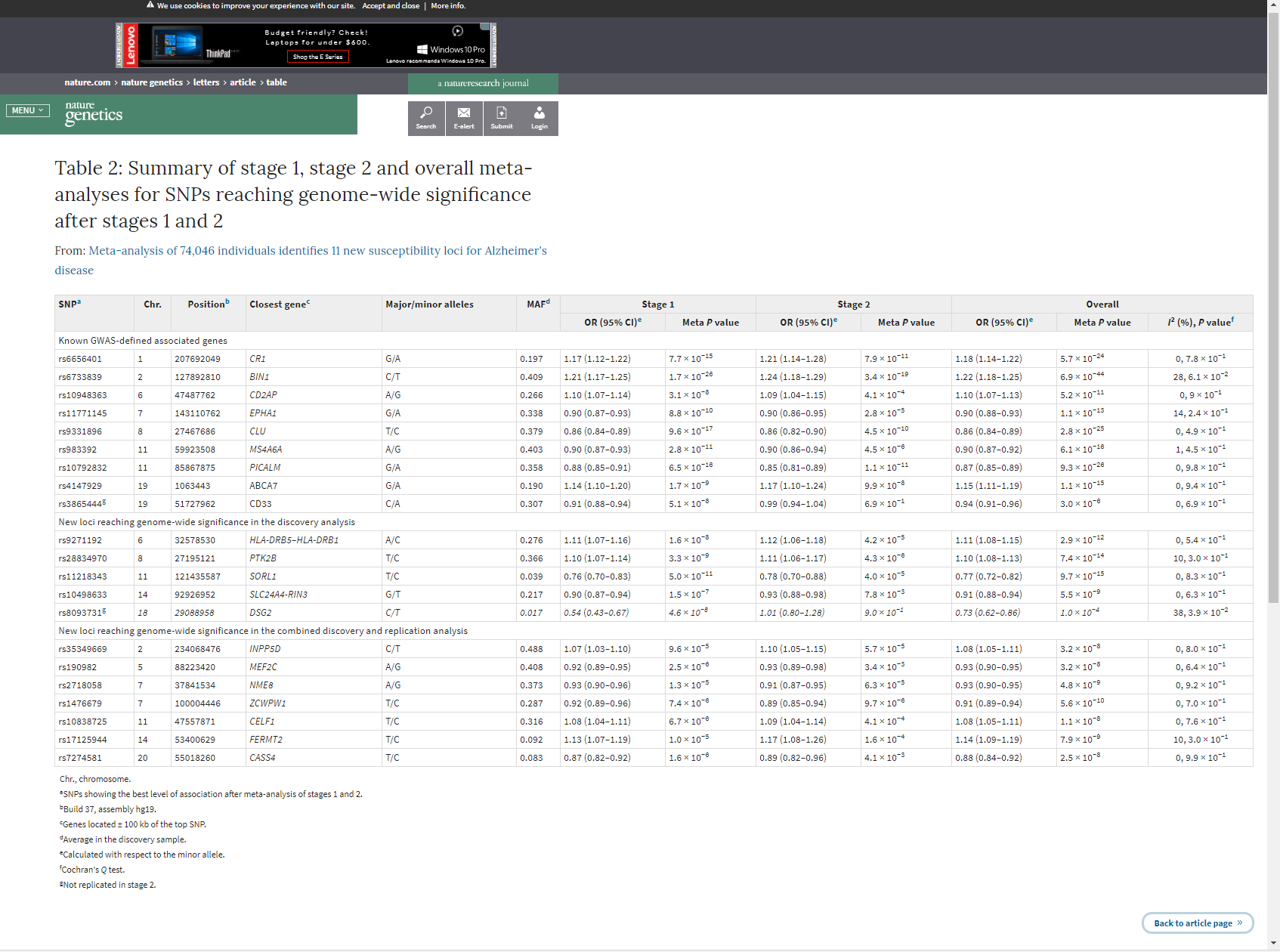
Example 1A: Scraping a html table from a webpage
Sometimes a table is online as part of a research article but can’t be easily coerced into a useful format. You can’t copy and paste the table into excel and its not stored elsewhere. In these situations you can use the handy R package rvest to scrape it into R from the webpage.
First load the rvest package to scrape the table.
library("rvest")
library("knitr")Next, get the url of the webpage where the table is stored.
url <- "https://www.nature.com/articles/ng.2802/tables/2"Now, the trickiest part of the process. We need find where the table lives on this webpage. An excellent guide to finding out how to do this can be found on Cory Nissen’s blogpost– this is also where I learned about using rvest to scrape html tables. Once you have copied the table location, the rest is easy!
table_path='//*[@id="content"]/div/div/figure/div[1]/div/div[1]/table'
nature_genetics_table2 <- url %>%
read_html() %>%
html_nodes(xpath=table_path) %>%
html_table(fill=T)
nature_genetics_table2 = nature_genetics_table2[[1]]This is what the first few lines of our scraped product looks like:
kable(nature_genetics_table2[1:4,])| SNPa | Chr. | Positionb | Closest genec | Major/minor alleles | MAFd | Stage 1 | Stage 1 | Stage 2 | Stage 2 | Overall | Overall | Overall |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNPa | Chr. | Positionb | Closest genec | Major/minor alleles | MAFd | OR (95% CI)e | Meta P value | OR (95% CI)e | Meta P value | OR (95% CI)e | Meta P value | I2 (%), P valuef |
| Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes | Known GWAS-defined associated genes |
| rs6656401 | 1 | 207692049 | CR1 | G/A | 0.197 | 1.17 (1.12–1.22) | 7.7 × 10−15 | 1.21 (1.14–1.28) | 7.9 × 10−11 | 1.18 (1.14–1.22) | 5.7 × 10−24 | 0, 7.8 × 10−1 |
| rs6733839 | 2 | 127892810 | BIN1 | C/T | 0.409 | 1.21 (1.17–1.25) | 1.7 × 10−26 | 1.24 (1.18–1.29) | 3.4 × 10−19 | 1.22 (1.18–1.25) | 6.9 × 10−44 | 28, 6.1 × 10−2 |
While this table has the information we want, it is clearly still a mess. Which brings us to…
Example 1B: Making messy data useful
Fortunately, there is the right number of columns, but there are lines of text breaking the table and stretching through at least one row of the table. There are two others (not shown), so getting these rows into better data format will be our first task.
Cleaning up the rows
We could look at the table and see these lines are on rows 2, 12, and 18. But let’s do this instead using some R code. The trick here is to note that all the elements of these rows contain the exact same text.
v=which(apply(nature_genetics_table2,1, function(x) length(unique(unlist(x))) )==1)Great, now let’s get rid of these rows but retain the information. We are going to do this in three steps. First, we will split the data.frame into a list based on the location of these descriptions (rows 2,12, and 18). Then, we will clean this list up by keeping only the list elements with data. We will move the text taking up entire rows to an additional column titled “Description”. Lastly, we will concatenate this cleaned list back into a data.frame.
nature_genetics_table2_list = split(nature_genetics_table2, cumsum(1:nrow(nature_genetics_table2) %in% v))
nature_genetics_table2_list = lapply(nature_genetics_table2_list[2:4], function(y){ y$Description= unique(as.character(y[1,]) )
y[-1,] } )
nature_genetics_table2_clean = do.call("rbind",nature_genetics_table2_list)Now let’s look at our data.
kable(nature_genetics_table2_clean[1:3,])| SNPa | Chr. | Positionb | Closest genec | Major/minor alleles | MAFd | Stage 1 | Stage 1 | Stage 2 | Stage 2 | Overall | Overall | Overall | Description | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1.3 | rs6656401 | 1 | 207692049 | CR1 | G/A | 0.197 | 1.17 (1.12–1.22) | 7.7 × 10−15 | 1.21 (1.14–1.28) | 7.9 × 10−11 | 1.18 (1.14–1.22) | 5.7 × 10−24 | 0, 7.8 × 10−1 | Known GWAS-defined associated genes |
| 1.4 | rs6733839 | 2 | 127892810 | BIN1 | C/T | 0.409 | 1.21 (1.17–1.25) | 1.7 × 10−26 | 1.24 (1.18–1.29) | 3.4 × 10−19 | 1.22 (1.18–1.25) | 6.9 × 10−44 | 28, 6.1 × 10−2 | Known GWAS-defined associated genes |
| 1.5 | rs10948363 | 6 | 47487762 | CD2AP | A/G | 0.266 | 1.10 (1.07–1.14) | 3.1 × 10−8 | 1.09 (1.04–1.15) | 4.1 × 10−4 | 1.10 (1.07–1.13) | 5.2 × 10−11 | 0, 9 × 10−1 | Known GWAS-defined associated genes |
Fixing column names
It’s getting better but is still messy. Let’s clean up those columns names. This part we will do by hand.
colnames(nature_genetics_table2_clean) <- c("SNP", "Chr", "Position", "Closest gene", "Major/minor alleles", "MAF", "Stage1_OR", "Stage1_MetaP", "Stage2_OR","Stage2_MetaP", "Overall_OR", "Overall_MetaP", "I2_Percent/P","Description")
colnames(nature_genetics_table2_clean)## [1] "SNP" "Chr" "Position"
## [4] "Closest gene" "Major/minor alleles" "MAF"
## [7] "Stage1_OR" "Stage1_MetaP" "Stage2_OR"
## [10] "Stage2_MetaP" "Overall_OR" "Overall_MetaP"
## [13] "I2_Percent/P" "Description"Making a character variable into a numeric variable
It’s coming along.. Next, we need to make the numbers into, well numbers. This will be more useful in R than a character class of data. To do this, try using the as.numeric function as shown.
as.numeric(nature_genetics_table2_clean$Stage1_MetaP)## Warning: NAs introduced by coercion## [1] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NAIt doesn’t work because there are weird symbols that R doesn’t understand. Get rid of them with the gsub command and replace them an E (scientific notation).
nature_genetics_table2_clean$Stage1_MetaP = gsub(" × 10","E",nature_genetics_table2_clean$Stage1_MetaP)Now try converting to a numeric.
as.numeric(nature_genetics_table2_clean$Stage1_MetaP)## Warning: NAs introduced by coercion## [1] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NAIt still doesn’t work!! Take a second closer look at the data. Can you discern why the code failed?
nature_genetics_table2_clean$Stage1_MetaP## [1] "7.7E-15" "1.7E-26" "3.1E-8" "8.8E-10" "9.6E-17" "2.8E-11" "6.5E-16"
## [8] "1.7E-9" "5.1E-8" "1.6E-8" "3.3E-9" "5.0E-11" "1.5E-7" "4.6E-8"
## [15] "9.6E-5" "2.5E-6" "1.3E-5" "7.4E-6" "6.7E-6" "1.0E-5" "1.6E-6"Yep, that’s right the − symbol is not in fact the same as a minus symbol -! We need to replace it. We’ll use the fact that that symbol always appears immediately after a capital E to our advantage.
Split the string on the E using strsplit.
s = strsplit(nature_genetics_table2_clean$Stage1_MetaP, "E")Get the first and second half of each string.
firstStr = lapply(s, `[[`, 1 )
secondStr=lapply(s, `[[`, 2 )Knock off that first character: which the symbol we don’t want and slap a minus sign back on .
secondStr= lapply(secondStr,function(x) {paste0("E-",substring(x,2))})Finally, stitch the two parts of the string back together now that the minus sign has been corrected and convert it to numeric.
mapply( function(firstStr, secondStr) {as.numeric(paste0(firstStr,secondStr) )}, firstStr, secondStr )## [1] 7.7e-15 1.7e-26 3.1e-08 8.8e-10 9.6e-17 2.8e-11 6.5e-16 1.7e-09
## [9] 5.1e-08 1.6e-08 3.3e-09 5.0e-11 1.5e-07 4.6e-08 9.6e-05 2.5e-06
## [17] 1.3e-05 7.4e-06 6.7e-06 1.0e-05 1.6e-06It works! Make sure to replace the column in the data.frame.
nature_genetics_table2_clean$Stage1_MetaP= mapply( function(firstStr, secondStr) {as.numeric(paste0(firstStr,secondStr) )}, firstStr, secondStr )See how it appears in the table now as a numeric? Try wrangling the rest of these columns into a useful format on your own and let me know how it goes.
kable(nature_genetics_table2_clean[1:3,])| SNP | Chr | Position | Closest gene | Major/minor alleles | MAF | Stage1_OR | Stage1_MetaP | Stage2_OR | Stage2_MetaP | Overall_OR | Overall_MetaP | I2_Percent/P | Description | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1.3 | rs6656401 | 1 | 207692049 | CR1 | G/A | 0.197 | 1.17 (1.12–1.22) | 0 | 1.21 (1.14–1.28) | 7.9 × 10−11 | 1.18 (1.14–1.22) | 5.7 × 10−24 | 0, 7.8 × 10−1 | Known GWAS-defined associated genes |
| 1.4 | rs6733839 | 2 | 127892810 | BIN1 | C/T | 0.409 | 1.21 (1.17–1.25) | 0 | 1.24 (1.18–1.29) | 3.4 × 10−19 | 1.22 (1.18–1.25) | 6.9 × 10−44 | 28, 6.1 × 10−2 | Known GWAS-defined associated genes |
| 1.5 | rs10948363 | 6 | 47487762 | CD2AP | A/G | 0.266 | 1.10 (1.07–1.14) | 0 | 1.09 (1.04–1.15) | 4.1 × 10−4 | 1.10 (1.07–1.13) | 5.2 × 10−11 | 0, 9 × 10−1 | Known GWAS-defined associated genes |
Conclusions
Today we went through getting data directly into R from an html table (a table on a webpage) and demonstrated how to get the data into a useful format. There are a couple advantages of doing this work in R instead of using excel– if that’s even an option. First, R is more reproducible. Second, once you have code for wrangling one table, wrangling the next one will be much faster. At the end of the day though, it is always easiest when the table is shared as a csv or excel file; something to keep in mind when preparing your own papers.
Reproducibility
## Session info ----------------------------------------------------------------------------------------------------------## setting value
## version R version 3.4.4 (2018-03-15)
## system x86_64, mingw32
## ui RTerm
## language (EN)
## collate English_United States.1252
## tz America/New_York
## date 2018-04-20## Packages --------------------------------------------------------------------------------------------------------------## package * version date source
## backports 1.1.2 2017-12-13 CRAN (R 3.4.3)
## base * 3.4.4 2018-03-15 local
## blogdown 0.5.12 2018-03-23 Github (rstudio/blogdown@21f14af)
## bookdown 0.7 2018-02-18 CRAN (R 3.4.3)
## compiler 3.4.4 2018-03-15 local
## curl 3.1 2017-12-12 CRAN (R 3.4.3)
## datasets * 3.4.4 2018-03-15 local
## devtools * 1.13.5 2018-02-18 CRAN (R 3.4.3)
## digest 0.6.15 2018-01-28 CRAN (R 3.4.3)
## evaluate 0.10.1 2017-06-24 CRAN (R 3.4.3)
## graphics * 3.4.4 2018-03-15 local
## grDevices * 3.4.4 2018-03-15 local
## highr 0.6 2016-05-09 CRAN (R 3.4.3)
## htmltools 0.3.6 2017-04-28 CRAN (R 3.4.3)
## httr 1.3.1 2017-08-20 CRAN (R 3.4.3)
## knitr * 1.20 2018-02-20 CRAN (R 3.4.3)
## magrittr 1.5 2014-11-22 CRAN (R 3.4.3)
## memoise 1.1.0 2017-04-21 CRAN (R 3.4.3)
## methods * 3.4.4 2018-03-15 local
## R6 2.2.2 2017-06-17 CRAN (R 3.4.3)
## Rcpp 0.12.16 2018-03-13 CRAN (R 3.4.4)
## rmarkdown 1.9 2018-03-01 CRAN (R 3.4.3)
## rprojroot 1.3-2 2018-01-03 CRAN (R 3.4.3)
## rvest * 0.3.2 2016-06-17 CRAN (R 3.4.3)
## selectr 0.3-2 2018-03-05 CRAN (R 3.4.3)
## stats * 3.4.4 2018-03-15 local
## stringi 1.1.7 2018-03-12 CRAN (R 3.4.4)
## stringr 1.3.0 2018-02-19 CRAN (R 3.4.3)
## tools 3.4.4 2018-03-15 local
## utils * 3.4.4 2018-03-15 local
## withr 2.1.2 2018-03-15 CRAN (R 3.4.4)
## xfun 0.1 2018-01-22 CRAN (R 3.4.3)
## xml2 * 1.2.0 2018-01-24 CRAN (R 3.4.3)
## yaml 2.1.18 2018-03-08 CRAN (R 3.4.3)