Welcome to the smokingMouse project! Here you’ll be able to access the mouse expression data used for the analysis of the smoking-nicotine-mouse LIBD project.
Overview
This bulk RNA-sequencing project consisted of a differential expression analysis (DEA) involving 4 data types: genes, transcripts, exons, and exon-exon junctions. The main goal of this study was to explore the effects of prenatal exposure to smoking and nicotine on the developing mouse brain. As secondary objectives, this work evaluated: 1) the affected genes by each exposure in the adult female brain in order to compare offspring and adult results, and 2) the effects of smoking on adult blood and brain to search for overlapping biomarkers in both tissues. Finally, DEGs identified in mouse were compared against previously published results in human (Semick et al., 2020 and Toikumo et al., 2023).
Study design
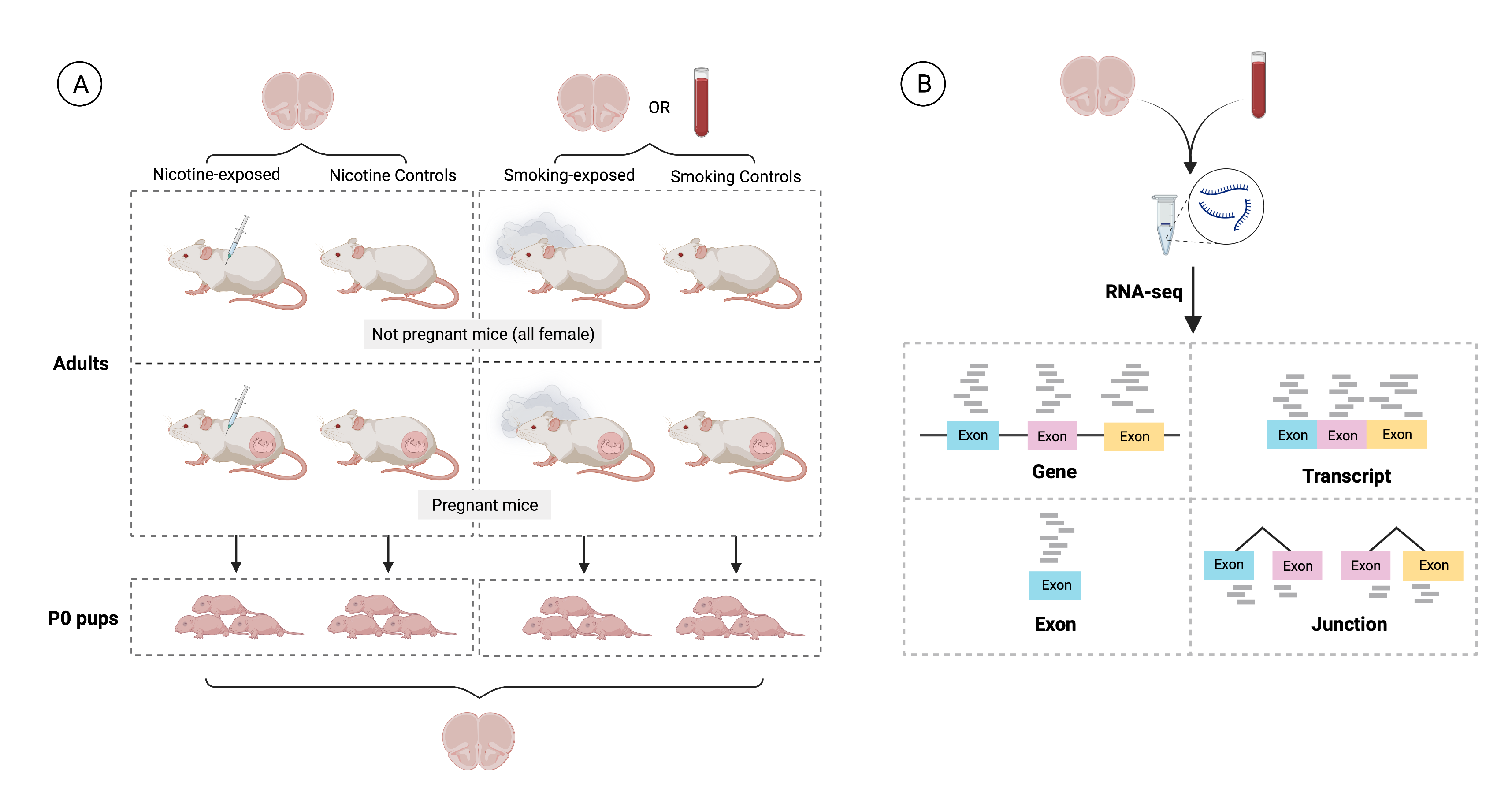
Experimental design of the study. A) 21 pregnant mice were split into two experiments: in the first one prenatal nicotine exposure (PNE) was modeled administering nicotine (n=3) or vehicle (n=3) to the dams during gestation, and in the second maternal smoking during pregnancy (MSDP) was modeled exposing dams to cigarette smoke during gestation (n=8) or using them as controls (n=7). A total of 137 pups were born: 19 were born to nicotine-administered mice, 23 to vehicle-administered mice, 46 to smoking-exposed mice, and 49 to smoking control mice. 17 nonpregnant adult females were also nicotine-administered (n=9) or vehicle-administered (n=8) to model adult nicotine exposure, and 9 additional nonpregnant dams were smoking-exposed (n=4) or controls (n=5) to model adult smoking. Frontal cortex samples of all P0 pups (n=137: 42 for PNE and 95 for MSDP) and adults (n=47: 23 for the nicotine experiment and 24 for the smoking experiment) were obtained, as well as blood samples from the smoking-exposed and smoking control adults (n=24), totaling 208 samples. Number of donors and samples are indicated in the figure. B) RNA was extracted from such samples and bulk RNA-seq experiments were performed, obtaining expression counts for genes, transcripts, exons, and exon-exon junctions.
Workflow
The next table summarizes the analyses done at each level.
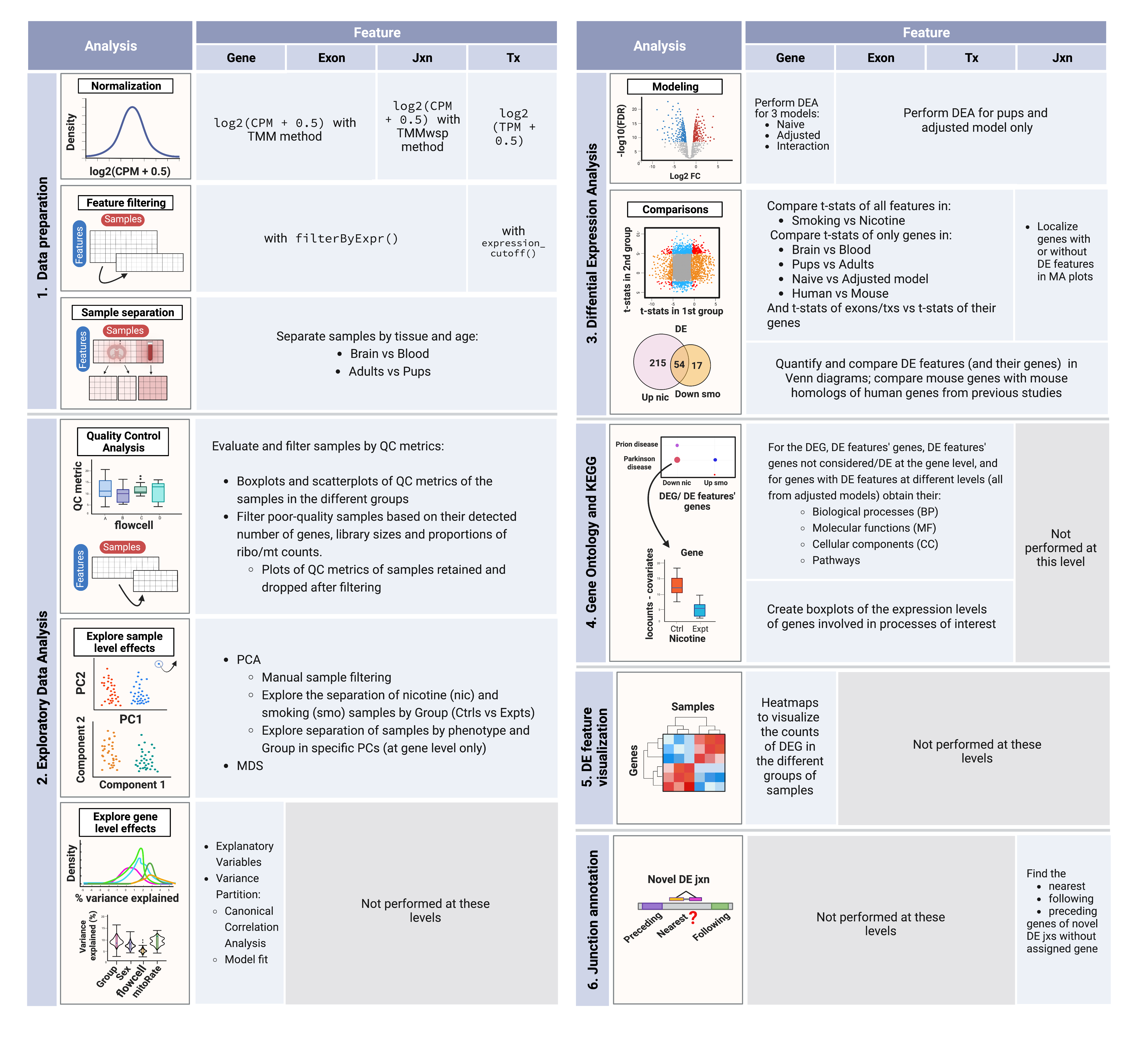
Summary of analysis steps across gene expression feature levels:
1. Data processing: counts of genes, exons, and exon-exon junctions were normalized to CPM and log2-transformed; transcript expression values were only log2-transformed since they were already in TPM. Lowly-expressed features were removed using the indicated functions and samples were separated by tissue and age in order to create subsets of the data for downstream analyses.
2. Exploratory Data Analysis (EDA): QC metrics of the samples were examined and used to filter the poor quality ones. Sample level effects were explored through dimensionality reduction methods and segregated samples in PCA plots were removed from the datasets. Gene level effects were evaluated with analyses of variance partition.
3. Differential Expression Analysis (DEA): with the relevant variables identified in the previous steps, the DEA was performed at the gene level for nicotine and smoking exposure in adult and pup brain samples, and for smoking exposure in adult blood samples; DEA at the rest of the levels was performed for both exposures in pup brain only. DE signals of the genes across the different experiments, sexes, ages, tissues, and species (using human results from Semick et al., 2020) were contrasted, as well as the DE signals of exons and transcripts vs those of their genes. Resultant DEGs and DE features (and their genes) were compared by direction of regulation (up/down) between and within exposures (nicotine/smoking); DEGs for smoking and nicotine exposure were compared between sexes and contrasted against sex-related DEGs in pup brain, and against human genes associated with TUD from Toikumo et al., 2023.
4. Functional Enrichment Analysis: GO & KEGG terms significantly enriched in the clusters of DEGs and genes of DE transcripts and exons were obtained.
5. DGE visualization: log2-normalized expression of DEGs was scaled and groups of up- and down-regulated genes were distinguished.
6. Novel junction gene annotation: for uncharacterized DE junctions with no annotated gene, their nearest, preceding, and following genes were determined.
Abbreviations: Jxn: junction; Tx(s): transcript(s); CPM: counts per million; TPM: transcripts per million; TMM: Trimmed Mean of M-Values; TMMwsp: TMM with singleton pairing; QC: quality control; PC: principal component; DEA: differential expression analysis; DE: differential expression/differentially expressed; FC: fold-change; FDR: false discovery rate; DEGs: differentially expressed genes; TUD: tobacco use disorder; DGE: differential gene expression.
All R scripts created to perform such analyses can be found in code on GitHub.
smoking Mouse datasets
The mouse datasets contain the following data in a single R RangedSummarizedExperiment* object for each feature (genes, transcripts, exons, and exon-exon junctions):
- Raw data: raw read counts (for genes, exons, and junctions) or TPM (for transcripts), also including the original metadata of the expression features and samples.
- Processed data: normalized and log-scaled counts of the same features (log(CPM+0.5) for genes, exons, and junctions, or log(TPM+0.5) for transcripts). In addition to the feature and sample information, the datasets contain information of which ones were used in downstream analyses (the ones that passed filtering steps), and which features were differentially expressed in the different experiments.
Moreover, you can find human data generated in Semick et al., (2018) in Mol Psychiatry (DOI: https://doi.org/10.1038/s41380-018-0223-1) that contain the results of a DEA for cigarette smoke exposure in adult and prenatal human brain.
*For more details, check the documentation for RangedSummarizedExperiment objects.
Data specifics
-
‘rse_gene_mouse_RNAseq_nic-smo.Rdata’: (
rse_geneobject) the gene RSE object contains the raw and log-normalized expression data of 55,401 mouse genes across the 208 samples from brain and blood of control and nicotine/smoking-exposed pup and adult mice. -
‘rse_tx_mouse_RNAseq_nic-smo.Rdata’: (
rse_txobject) the tx RSE object contains the raw and log-scaled expression data of 142,604 mouse transcripts across the 208 samples from brain and blood of control and nicotine/smoking-exposed pup and adult mice. -
‘rse_exon_mouse_RNAseq_nic-smo.Rdata’: (
rse_exonobject) the exon RSE object contains the raw and log-normalized expression data of 447,670 mouse exons across the 208 samples from brain and blood of control and nicotine/smoking-exposed pup and adult mice. -
‘rse_jx_mouse_RNAseq_nic-smo.Rdata’: (
rse_jxobject) the jx RSE object contains the raw and log-normalized expression data of 1,436,068 mouse exon-exon junctions across the 208 samples from brain and blood of control and nicotine/smoking-exposed pup and adult mice.
All the above datasets contain the sample and feature metadata and additional data of the results obtained in the filtering steps and the DEA.
-
‘de_genes_prenatal_human_brain_smoking.Rdata’: (
de_genes_prenatal_human_brain_smokingobject) data frame with DE statistics of 18,067 human genes for cigarette smoke exposure in prenatal human cortical tissue. -
‘de_genes_adult_human_brain_smoking.Rdata’: (
de_genes_adult_human_brain_smokingobject) data frame with DE statistics of 18,067 human genes for cigarette smoke exposure in adult human cortical tissue.
Installation instructions
Get the latest stable R release from CRAN. Then install smokingMouse from Bioconductor using the following code:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("smokingMouse")And the development version from GitHub with:
BiocManager::install("LieberInstitute/smokingMouse")Example of how to access the data
Below there’s example code on how to access the mouse and human gene data but can do the same for any of the datasets previously described. The datasets are retrieved from Bioconductor ExperimentHub.
## Connect to ExperimentHub
library(ExperimentHub)
#> Loading required package: BiocGenerics
#> Loading required package: generics
#>
#> Attaching package: 'generics'
#> The following objects are masked from 'package:base':
#>
#> as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
#> setequal, union
#>
#> Attaching package: 'BiocGenerics'
#> The following objects are masked from 'package:stats':
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from 'package:base':
#>
#> anyDuplicated, aperm, append, as.data.frame, basename, cbind,
#> colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
#> get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
#> order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
#> rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
#> unsplit, which.max, which.min
#> Loading required package: AnnotationHub
#> Loading required package: BiocFileCache
#> Loading required package: dbplyr
eh <- ExperimentHub::ExperimentHub()
#> snapshotDate(): 2024-11-13
## Load the datasets of the package
myfiles <- query(eh, "smokingMouse")
########################
# Mouse data
########################
## Download the mouse gene data
rse_gene <- myfiles[['EH8313']]
## This is a RangedSummarizedExperiment object
rse_gene
#> class: RangedSummarizedExperiment
#> dim: 55401 208
#> metadata(1): Obtained_from
#> assays(2): counts logcounts
#> rownames(55401): ENSMUSG00000102693.1 ENSMUSG00000064842.1 ...
#> ENSMUSG00000064371.1 ENSMUSG00000064372.1
#> rowData names(13): Length gencodeID ... DE_in_pup_brain_nicotine
#> DE_in_pup_brain_smoking
#> colnames: NULL
#> colData names(71): SAMPLE_ID FQCbasicStats ...
#> retained_after_QC_sample_filtering
#> retained_after_manual_sample_filtering
## Check sample info
colData(rse_gene)[1:5, 1:5]
#> DataFrame with 5 rows and 5 columns
#> SAMPLE_ID FQCbasicStats perBaseQual perTileQual perSeqQual
#> <character> <character> <character> <character> <character>
#> 1 Sample_2914 PASS PASS PASS PASS
#> 2 Sample_4042 PASS PASS PASS PASS
#> 3 Sample_4043 PASS PASS PASS PASS
#> 4 Sample_4044 PASS PASS PASS PASS
#> 5 Sample_4045 PASS PASS PASS PASS
## Check gene info
rowData(rse_gene)[1:5, 1:5]
#> DataFrame with 5 rows and 5 columns
#> Length gencodeID ensemblID
#> <integer> <character> <character>
#> ENSMUSG00000102693.1 1070 ENSMUSG00000102693.1 ENSMUSG00000102693
#> ENSMUSG00000064842.1 110 ENSMUSG00000064842.1 ENSMUSG00000064842
#> ENSMUSG00000051951.5 6094 ENSMUSG00000051951.5 ENSMUSG00000051951
#> ENSMUSG00000102851.1 480 ENSMUSG00000102851.1 ENSMUSG00000102851
#> ENSMUSG00000103377.1 2819 ENSMUSG00000103377.1 ENSMUSG00000103377
#> gene_type EntrezID
#> <character> <character>
#> ENSMUSG00000102693.1 TEC 71042
#> ENSMUSG00000064842.1 snRNA NA
#> ENSMUSG00000051951.5 protein_coding 497097
#> ENSMUSG00000102851.1 processed_pseudogene 100418032
#> ENSMUSG00000103377.1 TEC NA
## Access the original counts
original_counts <- assays(rse_gene)$counts
## Access the log-normalized counts
logcounts <- assays(rse_gene)$logcounts
########################
# Human data
########################
## Download the human gene data
de_genes_prenatal_human_brain_smoking <- myfiles[['EH8317']]
## This is a data frame
de_genes_prenatal_human_brain_smoking[1:5, ]
#> GRanges object with 5 ranges and 9 metadata columns:
#> seqnames ranges strand | Length Symbol
#> <Rle> <IRanges> <Rle> | <integer> <character>
#> ENSG00000080709 chr5 113696642-113832337 + | 3995 KCNN2
#> ENSG00000070886 chr1 22890057-22930087 + | 5358 EPHA8
#> ENSG00000218336 chr4 183065140-183724177 + | 11983 TENM3
#> ENSG00000189108 chrX 103810996-105011822 + | 3146 IL1RAPL2
#> ENSG00000186732 chr22 43807202-43903728 + | 5821 MPPED1
#> EntrezID logFC AveExpr t P.Value adj.P.Val
#> <integer> <numeric> <numeric> <numeric> <numeric> <numeric>
#> ENSG00000080709 3781 -0.694069 2.86444 -6.09779 2.59861e-06 0.0469491
#> ENSG00000070886 2046 1.545861 1.58351 5.67106 7.49034e-06 0.0477263
#> ENSG00000218336 55714 0.804367 6.31125 5.55661 9.97733e-06 0.0477263
#> ENSG00000189108 26280 -1.035988 1.62624 -5.53375 1.05665e-05 0.0477263
#> ENSG00000186732 758 0.384536 9.34706 5.41518 1.42396e-05 0.0514535
#> B
#> <numeric>
#> ENSG00000080709 4.44638
#> ENSG00000070886 3.18385
#> ENSG00000218336 3.53830
#> ENSG00000189108 2.83949
#> ENSG00000186732 3.19865
#> -------
#> seqinfo: 25 sequences from an unspecified genome; no seqlengths
## Access data of human genes as normally do with data framesCitation
Below is the citation output from using citation('smokingMouse') in R. Please run this yourself to check for any updates on how to cite smokingMouse.
print(citation('smokingMouse'), bibtex = TRUE)
#> To cite package 'smokingMouse' in publications use:
#>
#> Gonzalez-Padilla D, Collado-Torres L (2024). _Provides access to
#> smokingMouse project data_. doi:10.18129/B9.bioc.smokingMouse
#> <https://doi.org/10.18129/B9.bioc.smokingMouse>,
#> https://github.com/LieberInstitute/smokingMouse/smokingMouse - R
#> package version 1.5.1,
#> <http://www.bioconductor.org/packages/smokingMouse>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Manual{,
#> title = {Provides access to smokingMouse project data},
#> author = {Daianna Gonzalez-Padilla and Leonardo Collado-Torres},
#> year = {2024},
#> url = {http://www.bioconductor.org/packages/smokingMouse},
#> note = {https://github.com/LieberInstitute/smokingMouse/smokingMouse - R package version 1.5.1},
#> doi = {10.18129/B9.bioc.smokingMouse},
#> }
#>
#> Gonzalez-Padilla D, Eagles NJ, Cano M, Pertea G, Jaffe AE, Maynard
#> KR, Hancock DB, Handa JT, Martinowich K, Maynard KR, Collado-Torres L
#> (2024). "Molecular impact of nicotine and smoking exposure on the
#> developing and adult mouse brain." _bioRxiv_.
#> doi:10.1101/2024.11.05.622149
#> <https://doi.org/10.1101/2024.11.05.622149>,
#> <https://doi.org/10.1101/2024.11.05.622149>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Article{,
#> title = {Molecular impact of nicotine and smoking exposure on the developing and adult mouse brain},
#> author = {Daianna Gonzalez-Padilla and Nicholas J. Eagles and Marisol Cano and Geo Pertea and Andrew E. Jaffe and Kristen R. Maynard and Dana B. Hancock and James T. Handa and Keri Martinowich and Kristen R. Maynard and Leonardo Collado-Torres},
#> year = {2024},
#> journal = {bioRxiv},
#> doi = {10.1101/2024.11.05.622149},
#> url = {https://doi.org/10.1101/2024.11.05.622149},
#> }
#>
#>
#> To cite the original smoking-nicotine mouse work please use:
#>
#> (TODO)
#>
#>
#> To cite the original work from which human data come please use the following citation:
#>
#> Semick, S. A., Collado-Torres, L., Markunas, C. A., Shin, J. H., Deep-Soboslay, A., Tao, R., ...
#> & Jaffe, A. E. (2020). Developmental effects of maternal smoking during pregnancy on the human
#> frontal cortex transcriptome. Molecular psychiatry, 25(12), 3267-3277.
#> Please note that the smokingMouse package and the study analyses were only made possible thanks to many other R and bioinformatics software authors, which are cited either in the vignette and/or the paper describing this study.
Code of Conduct
Please note that the smokingMouse project is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.
Development tools
- Continuous code testing is possible thanks to GitHub actions through usethis, remotes, and rcmdcheck customized to use Bioconductor’s docker containers and BiocCheck.
- Code coverage assessment is possible thanks to codecov and covr.
- The documentation website is automatically updated thanks to pkgdown.
- The code is styled automatically thanks to styler.
- The documentation is formatted thanks to devtools and roxygen2.
For more details, check the dev directory.
This package was developed using biocthis.







