7 Outputs
7.1 Main Outputs
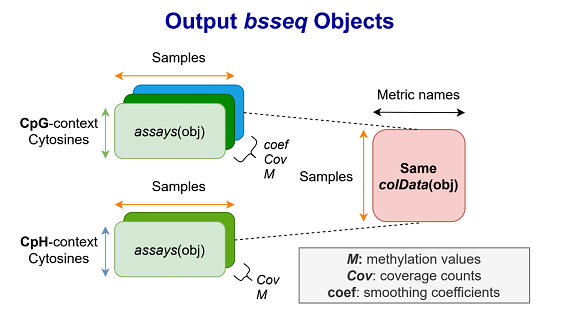
The major outputs from the second module are R objects from the bsseq Bioconductor package, which contain methylation proportion and coverage information at all cytosine loci in the human genome. bsseq extends the SummarizedExperiment class, which provides a general and popular format for storing genomics data. Two bsseq objects are produced, with one object containing cytosine sites in CpG context, and the other containing the remaining CpH loci.
7.1.1 CpG bsseq object
We “strand collapse” CpG loci, which involves combining methylation data from both genomic strands (and thus discarding strand-specific information). The object is “smoothed” with the BSmooth algorithm, a process for inferring stable regional estimates of methylation levels. Loci are ordered by genomic position.
7.1.2 CpH bsseq object
We retain strand-specific information for CpH loci. These loci are also ordered by genomic position.
7.1.3 Storage method
Because all cytosines in the genome are included in the output objects, the data may occupy tens or even hundreds of gigabytes in memory (RAM) if loaded in a typical fashion. To enable working with the objects in a reasonable amount of memory, the assays (in this case methylation fraction and coverage counts) are HDF5-backed using the HDF5Array R package, based on the DelayedArray framework. Essentially, this involves storing the assays in a .h5 file, a format designed to enable working with on-disk data as if it were loaded in RAM.
7.1.4 Metrics
The colData slot of each bsseq object includes metrics collected from various processing steps throughout the pipeline. These metrics are also included in a standalone RDA file containing an R data frame whose columns are different quality metrics, and whose rows are associated with sample names. A list of the exact column names and their descriptions is given below.
| Metric Name | Processing step | Description |
|---|---|---|
| FQCbasicStats | FastQC | Value for “Basic Statistics” in FastQC summary output |
| perBaseQual | FastQC | Value for “Per base sequence quality” in FastQC summary output |
| perTileQual | FastQC | Value for “Per tile sequence quality” in FastQC summary output |
| perSeqQual | FastQC | Value for “Per sequence quality scores” in FastQC summary output |
| perBaseContent | FastQC | Value for “Per base sequence content” in FastQC summary output |
| GCcontent | FastQC | Value for “Per sequence GC content” in FastQC summary output |
| Ncontent | FastQC | Value for “Per base N content” in FastQC summary output |
| SeqLengthDist | FastQC | Value for “Sequence Length Distribution” in FastQC summary output |
| SeqDuplication | FastQC | Value for “Sequence Duplication Levels” in FastQC summary output |
| OverrepSeqs | FastQC | Value for “Overrepresented sequences” in FastQC summary output |
| AdapterContent | FastQC | Value for “Adapter Content” in FastQC summary output |
| KmerContent | FastQC | Value for “Kmer Content” in FastQC summary output |
| Total_reads_processed | Trim Galore! | Value for “Total reads processed” entry in STDOUT from trimming |
| Reads_with_adapters | Trim Galore! | Value for “Reads with adapters” entry in STDOUT from trimming |
| Reads_written_passing_filters | Trim Galore! | Value for “Reads written (passing filters)” entry in STDOUT from trimming |
| Total_basepairs_processed | Trim Galore! | Value for “Total basepairs processed” entry in STDOUT from trimming |
| Quality_trimmed | Trim Galore! | Value for “Quality-trimmed” entry in STDOUT from trimming |
| Total_written_filtered | Trim Galore! | Value for “Total written (filtered)” entry in STDOUT from trimming |
| Sequence | Trim Galore! | Detected adapter sequence when trimming |
| was_trimmed | Trim Galore! | Logical value (“TRUE” or “FALSE”) indicating whether this sample was trimmed |
| pairs | Arioc | (Paired-end only) Total number of pairs present in input data |
| conc_pairs_total | Arioc | (Paired-end only) Total number of concordantly aligned pairs |
| conc_pairs_1_mapping | Arioc | (Paired-end only) Number of uniquely mapped concordant alignments |
| conc_pairs_many_mappings | Arioc | (Paired-end only) Number of non-uniquely mapped concordant pairs |
| disc_pairs | Arioc | (Paired-end only) Number of pairs that aligned discordantly |
| rejected_pairs | Arioc | (Paired-end only) Number of pairs that were “rejected” during alignment |
| unmapped_pairs | Arioc | (Paired-end only) Number of pairs that didn’t both successfully map |
| mates_not_in_paired_maps_total | Arioc | (Paired-end only) Value for “mates not in paired mappings” field in “SAM output” section of verbose output from alignment |
| mates_NIPM_with_no_maps | Arioc | (Paired-end only) Number of mates not in paired mappings having no mappings |
| mates_NIPM_with_1_map | Arioc | (Paired-end only) Number of mates not in paired mappings having 1 mapping |
| mates_NIPM_with_many_maps | Arioc | (Paired-end only) Number of mates not in paired mappings having 2 or more mappings |
| total_mapped_mates | Arioc | (Paired-end only) Total number of mates mapped |
| maxQlen | Arioc | Value for “maximum Q length” field in “SAM output” section of verbose output from alignment |
| max_diag_band_width | Arioc | (Paired-end only) Value for “maximum diagonal band width” field in “SAM output” section of verbose output from alignment |
| TLEN_mean | Arioc | (Paired-end only) Mean fragment length |
| TLEN_sd | Arioc | (Paired-end only) Standard deviation of fragment length |
| TLEN_skewness | Arioc | (Paired-end only) Skewness of fragment length |
| reads | Arioc | (Single-end only) Total number of reads present in input data |
| mapped_reads_total | Arioc | (Single-end only) Number of reads successfully mapped to the reference genome |
| mapped_reads_1_map | Arioc | (Single-end only) Number of reads mapped to a unique location on the reference genome |
| mapped_reads_many_maps | Arioc | (Single-end only) Number of reads mapped to more than 1 location on the reference genome |
| unmapped_reads | Arioc | (Single-end only) Number of reads that didn’t successfully map to the reference genome |
| duplicate_maps | Arioc | Value for “duplicate mappings (unreported)” field in “SAM output” section of verbose output from alignment |
| err_rate_primary_maps | Arioc | Observed error rate for reads (single-end) or pairs (paired-end) in primary mappings |
| perc_M_CpG | Methylation extraction | Among all cytosines observed in CpG context, the percent that are methylated |
| perc_M_CHG | Methylation extraction | Among all cytosines observed in CHG context, the percent that are methylated |
| perc_M_CHH | Methylation extraction | Among all cytosines observed in CHH context, the percent that are methylated |
| lambda_bs_conv_eff | Lambda pseudo-alignment | Estimated bisulfite conversion efficiency as a percentage |
7.2 Intermediary Outputs
BiocMAP generates a number of files along the process before producing the main outputs of interest in each module. Each of these “intermediary” files is described below.
7.2.1 First Module
Preprocessing Logs preprocessing/
preprocess_input_first_half.log: Information about how the inputsamples.manifestfile was parsed to internally handle input files correctly downstream.
FastQC Outputs fastQC/
[trim_status]/[file_name]/*: Outputs from FastQC. Here[trim_status]indicates when FastQC was performed:Untrimmedis before trimming, andTrimmedis after.[file_name]contains the sample name, and if applicable, the mate number.
Trimmed FASTQ Files trimming/
[sample_name]_trimmed*.fastq: Trimmed FASTQ files, if applicable, from Trim Galore.
Raw Alignment-related Files Arioc/
sams/[sample_name].c.sam: Concordant alignments output directly from Arioc, in SAM format.configs/[sample_name]_align.cfg: The exact config file used by Arioc to align reads to the reference genome.configs/[sample_name]_encode.cfg: The exact config file used by Arioc to encode reads prior to alignment to the reference genome.logs/*_[sample_name].log: Logs from encoding and aligning samples with Arioc.
Filtered Alignments FilteredAlignments/bams
[sample_name].cfu.sorted.bamand[sample_name].cfu.sorted.bam.bai: Coordinate-sorted, quality-filtered (only including alignments withMAPQ>= 5), unique (duplicate mappings removed) alignments in BAM format, with a corresponding BAM index.
7.2.2 Second Module
Preprocessing Logs preprocessing/
preprocess_input_second_half.log: Information about how the inputrules.txtfile was parsed to internally handle input files correctly downstream.
Lambda-based Bisulfite-Conversion Estimatation lambda/
[sample_name]_lambda_pseudo.log: (For experiments with lambda spike-ins, using the--with_lambdaoption) Logs containing a percentage estimate for the bisulfite-conversion efficiency for each sample, based on pseudo-aligning input FASTQs to both the lambda bacteriophage transcriptome and an “in silico bisulfite-converted copy” of the transcriptome, and comparing alignment rates.
Bismark Methylation Extractor Outputs BME/
[sample_name]/*: (When using the--use_bmeoption) Files output from runningbismark_methylation_extractor, including compressed text-based methylation information, a splitting report, and an “M-bias” report.
Cytosine Reports and Bedgraphs Reports/
[sample_name]/[sample_name].*.CX_report.txt: “Cytosine reports” containing methylation counts and context for every cytosine in the genome, split by chromosome. By default, these are generated withMethylDackel, or otherwise withcoverage2cytosinefrom theBismarksoftware suite when using the--use_bmeoption.[sample_name]/[sample_name]_bedgraph_merged*.gz: (When using the--use_bmeoption) Abedgraphfile containing positional and methylation information for each cytosine; a “coverage” file containing similar information. See Bismark’s documentation for more detail.
Individual bsseq objects BSobjects/objects/
[chromosome]/[context]/*: R objects containing either cytosines in “CpG” or “CpH”, for a single chromosome. While the final bsseq objects are combined across all chromosomes for convenience, it may be more practical in some cases to work with these individual objects, which are significantly smaller in memory.