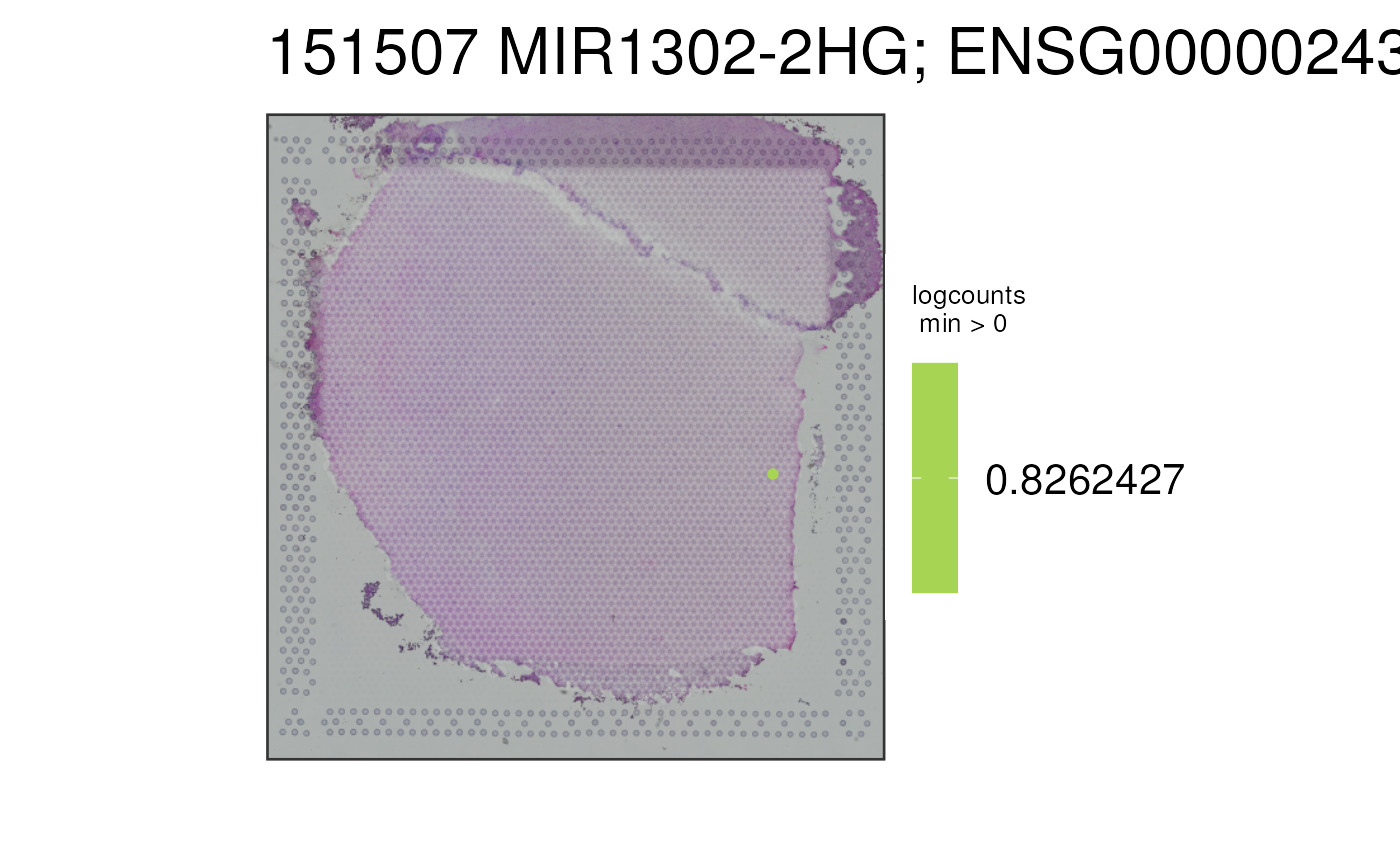
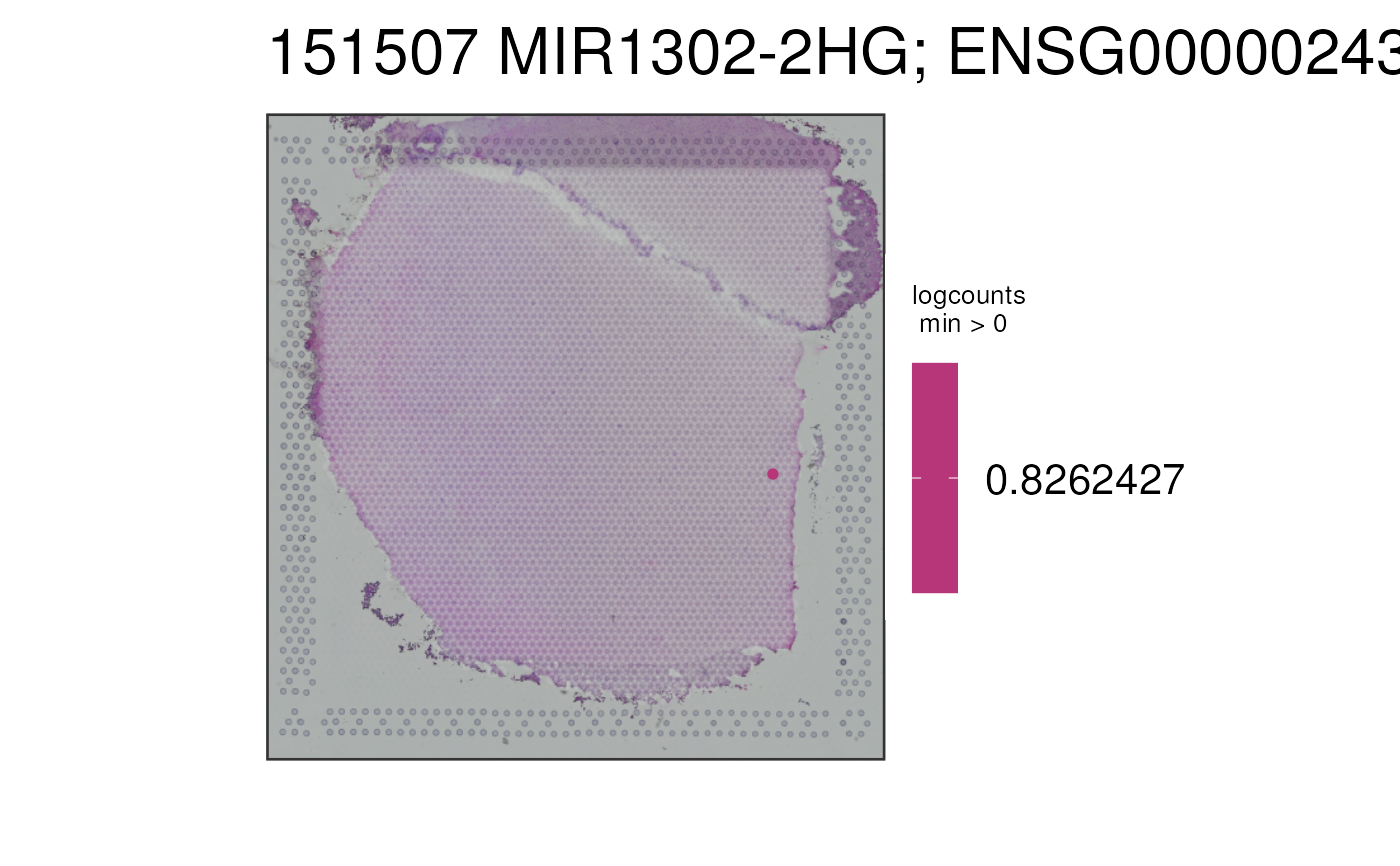
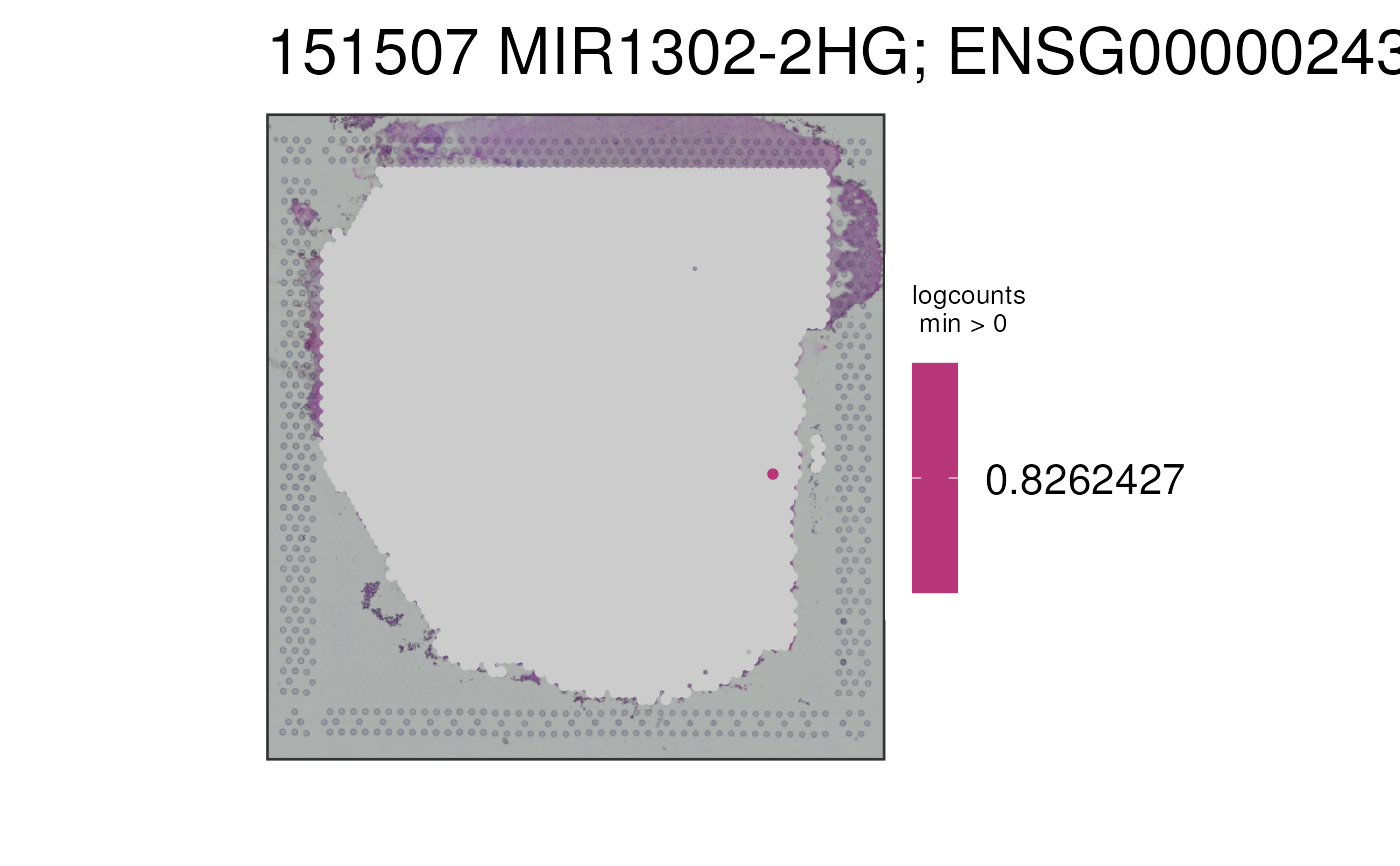
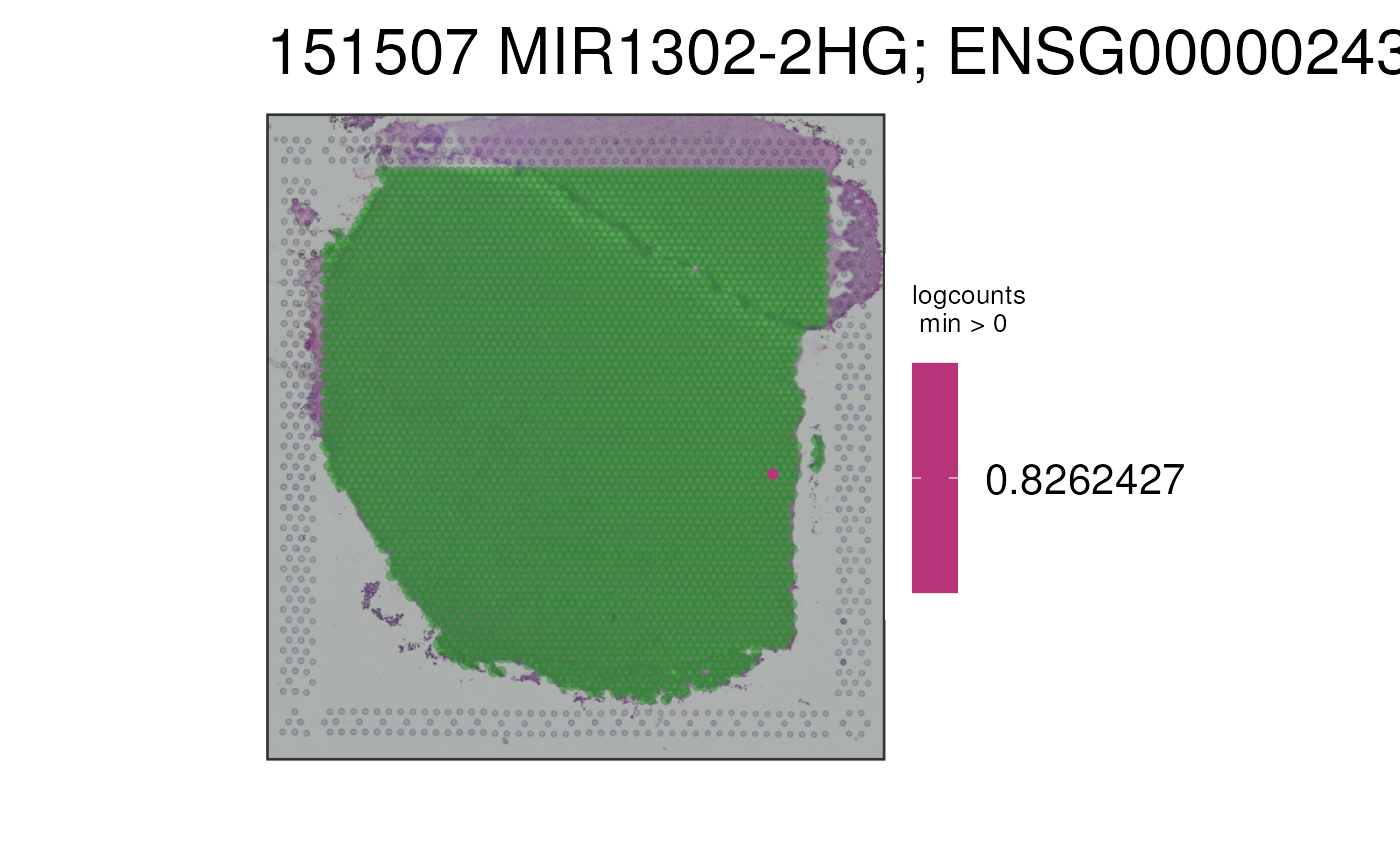
This function visualizes the gene expression stored in assays(spe) or any
continuous variable stored in colData(spe) for one given sample at the
spot-level using (by default) the histology information on the background.
To visualize clusters (or any discrete variable) use vis_clus().
vis_gene(
spe,
sampleid = unique(spe$sample_id)[1],
geneid = rowData(spe)$gene_search[1],
spatial = TRUE,
assayname = "logcounts",
minCount = 0,
viridis = TRUE,
image_id = "lowres",
alpha = NA,
cont_colors = if (viridis) viridisLite::viridis(21) else c("aquamarine4",
"springgreen", "goldenrod", "red"),
point_size = 2,
auto_crop = TRUE,
na_color = "#CCCCCC40",
multi_gene_method = c("z_score", "pca", "sparsity"),
is_stitched = FALSE,
cap_percentile = 1,
...
)Arguments
- spe
A SpatialExperiment-class object. See
fetch_data()for how to download some example objects orread10xVisiumWrapper()to read inspaceranger --countoutput files and build your ownspeobject.- sampleid
A
character(1)specifying which sample to plot fromcolData(spe)$sample_id(formerlycolData(spe)$sample_name).- geneid
A
character()specifying the gene ID(s) stored inrowData(spe)$gene_searchor a continuous variable(s) stored incolData(spe)to visualize. For each ID, ifrowData(spe)$gene_searchis missing, thenrownames(spe)is used to search for the gene ID. When a vector of length > 1 is supplied, the continuous variables are combined according tomulti_gene_method, producing a single value for each spot.- spatial
A
logical(1)indicating whether to include the histology layer fromgeom_spatial(). If you plan to use ggplotly() then it's best to set this toFALSE.- assayname
The name of the
assays(spe)to use for extracting the gene expression data. Defaults tologcounts.- minCount
A
numeric(1)specifying the minimum gene expression (or value in the continuous variable) to visualize. Values at or below this threshold will be set toNA. Defaults to0.- viridis
A
logical(1)whether to use the color-blind friendly palette from viridis or the color palette used in the paper that was chosen for contrast when visualizing the data on top of the histology image. One issue is being able to differentiate low values from NA ones due to the purple-ish histology information that is dependent on cell density.- image_id
A
character(1)with the name of the image ID you want to use in the background.- alpha
A
numeric(1)in the[0, 1]range that specifies the transparency level of the data on the spots.- cont_colors
A
character()vector of colors that supersedes theviridisargument.- point_size
A
numeric(1)specifying the size of the points. Defaults to1.25. Some colors look better if you use2for instance.- auto_crop
A
logical(1)indicating whether to automatically crop the image / plotting area, which is useful if the Visium capture area is not centered on the image and if the image is not a square.- na_color
A
character(1)specifying a color for the NA values. If you setalpha = NAthen it's best to setna_colorto a color that has alpha blending already, which will make non-NA values pop up more and the NA values will show with a lighter color. This behavior is lost whenalphais set to a non-NAvalue.- multi_gene_method
A
character(1): either"pca","sparsity", or"z_score". This parameter controls how multiple continuous variables are combined for visualization, and only applies whengeneidhas length great than 1.z_score: to summarize multiple continuous variables, each is normalized to represent a Z-score. The multiple scores are then averaged.pca: PCA dimension reduction is conducted on the matrix formed by the continuous variables, and the first PC is then used and multiplied by -1 if needed to have the majority of the values for PC1 to be positive.sparsity: the proportion of continuous variables with positive values for each spot is computed. For more details, check the multi gene vignette at https://research.libd.org/spatialLIBD/articles/multi_gene_plots.html.- is_stitched
A
logical(1)vector: IfTRUE, expects a SpatialExperiment-class built withvisiumStitched::build_spe(). http://research.libd.org/visiumStitched/reference/build_spe.html; in particular, expects a logical colData columnexclude_overlappingspecifying which spots to exclude from the plot. Setsauto_crop = FALSE.- cap_percentile
A
numeric(1)in (0, 1] determining the maximum percentile (as a proportion) at which to cap expression. For example, a value of 0.95 sets the top 5% of expression values to the 95th percentile value. This can help make the color scale more dynamic in the presence of high outliers. Defaults to1, which effectively performs no capping.- ...
Passed to paste0() for making the title of the plot following the
sampleid.
Value
A ggplot2 object.
Details
This function subsets spe to the given sample and prepares the
data and title for vis_gene_p(). It also adds a caption to the plot.
See also
Other Spatial gene visualization functions:
vis_gene_p(),
vis_grid_gene()
Examples
if (enough_ram()) {
## Obtain the necessary data
if (!exists("spe")) spe <- fetch_data("spe")
## Valid `geneid` values are those in
head(rowData(spe)$gene_search)
## or continuous variables stored in colData(spe)
## or rownames(spe)
## Visualize a default gene on the non-viridis scale
p1 <- vis_gene(
spe = spe,
sampleid = "151507",
viridis = FALSE
)
print(p1)
## Use a custom set of colors in the reverse order than usual
p2 <- vis_gene(
spe = spe,
sampleid = "151507",
cont_colors = rev(viridisLite::viridis(21, option = "magma"))
)
print(p2)
## Turn the alpha to 1, which makes the NA values have a full alpha
p2b <- vis_gene(
spe = spe,
sampleid = "151507",
cont_colors = rev(viridisLite::viridis(21, option = "magma")),
alpha = 1
)
print(p2b)
## Turn the alpha to NA, and use an alpha-blended "forestgreen" for
## the NA values
# https://gist.githubusercontent.com/mages/5339689/raw/2aaa482dfbbecbfcb726525a3d81661f9d802a8e/add.alpha.R
# add.alpha("forestgreen", 0.5)
p2c <- vis_gene(
spe = spe,
sampleid = "151507",
cont_colors = rev(viridisLite::viridis(21, option = "magma")),
alpha = NA,
na_color = "#228B2280"
)
print(p2c)
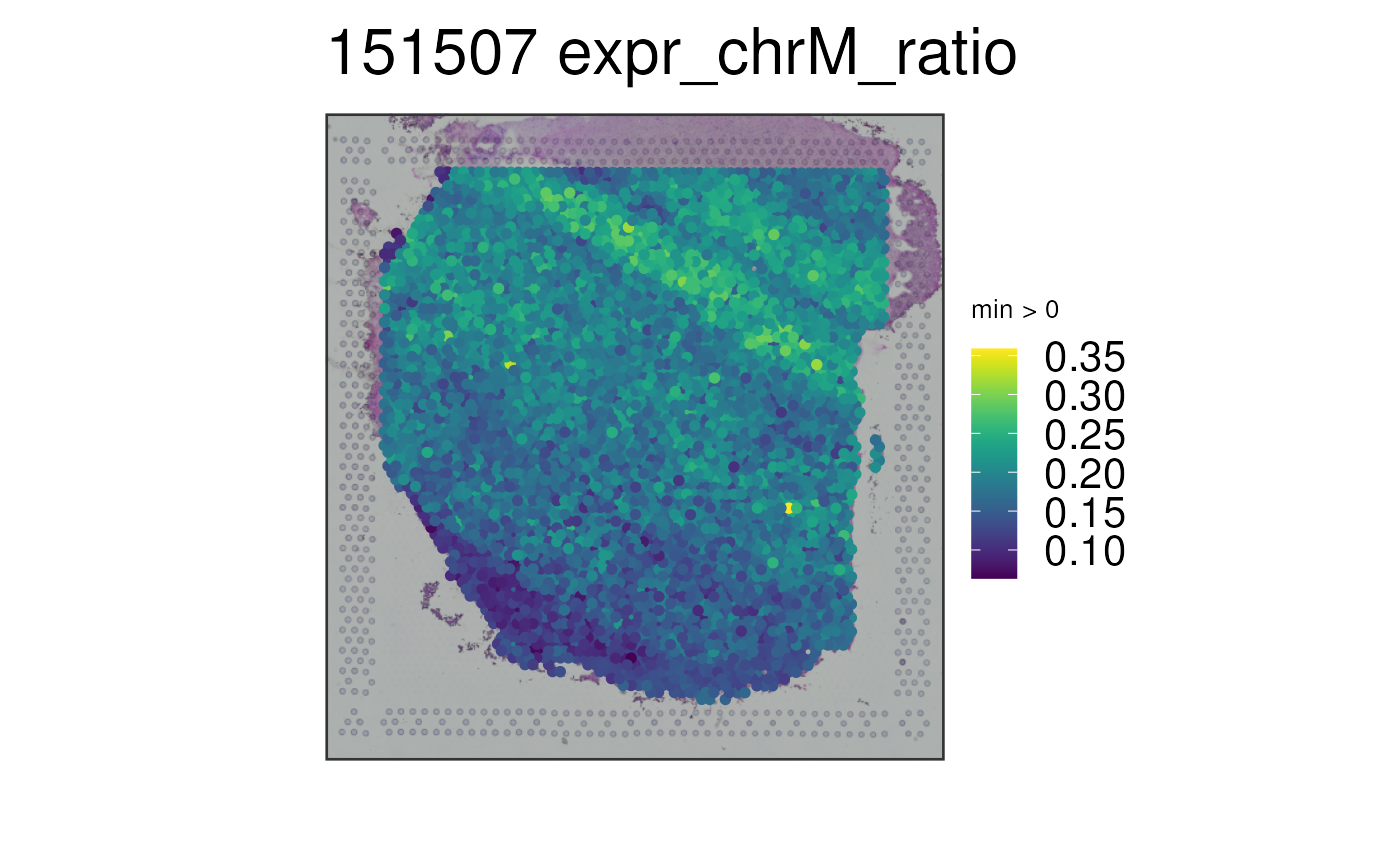
## Visualize a continuous variable, in this case, the ratio of chrM
## gene expression compared to the total expression at the spot-level
p3 <- vis_gene(
spe = spe,
sampleid = "151507",
geneid = "expr_chrM_ratio"
)
print(p3)
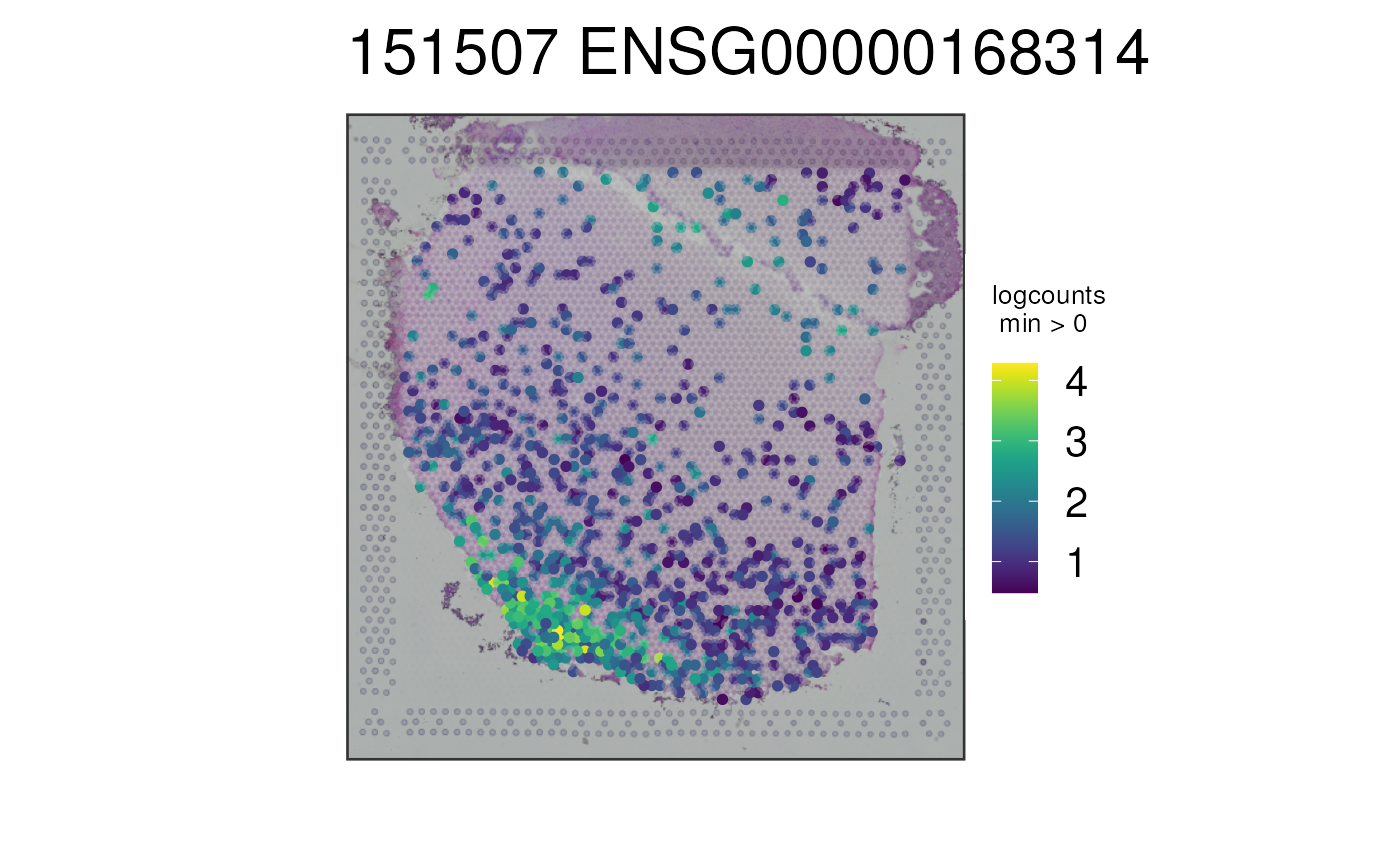
## Visualize a gene using the rownames(spe)
p4 <- vis_gene(
spe = spe,
sampleid = "151507",
geneid = rownames(spe)[which(rowData(spe)$gene_name == "MOBP")]
)
print(p4)
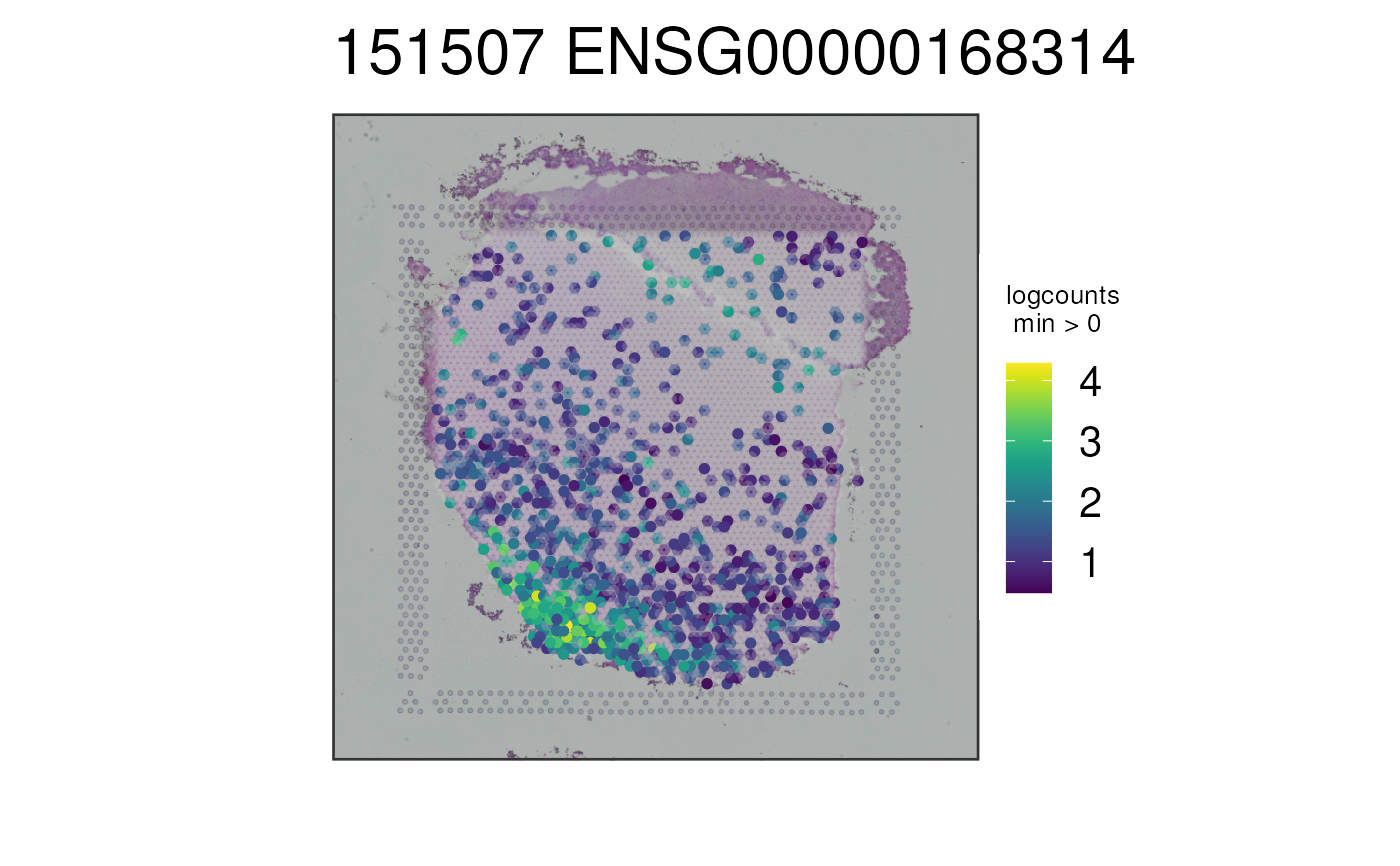
## Repeat without auto-cropping the image
p5 <- vis_gene(
spe = spe,
sampleid = "151507",
geneid = rownames(spe)[which(rowData(spe)$gene_name == "MOBP")],
auto_crop = FALSE
)
print(p5)
# Define several markers for white matter
white_matter_genes <- c(
"ENSG00000197971", "ENSG00000131095", "ENSG00000123560",
"ENSG00000171885"
)
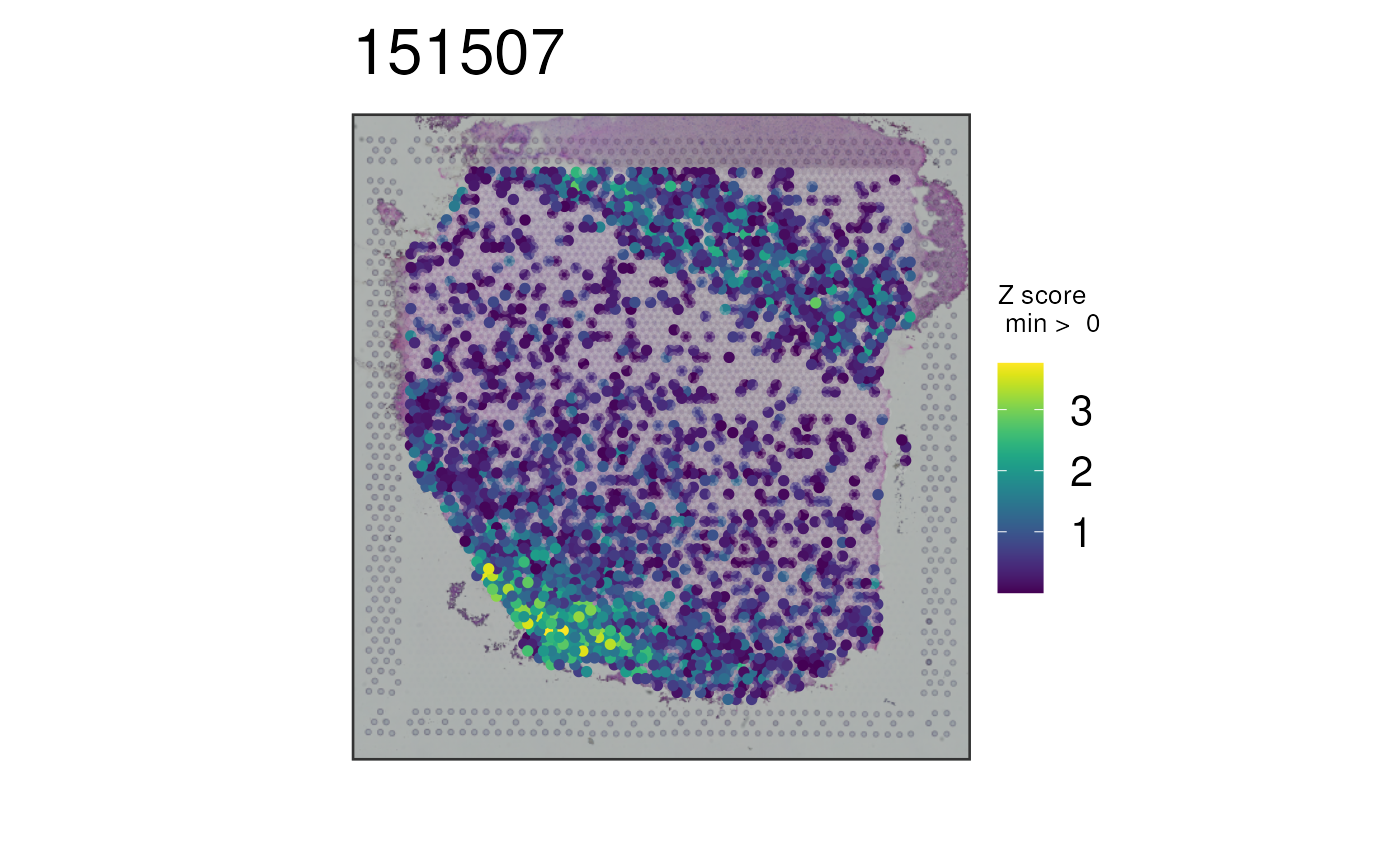
## Plot all white matter markers at once using the Z-score combination
## method. Flatten this quantity at the top 5% of values for plotting
p6 <- vis_gene(
spe = spe,
sampleid = "151507",
geneid = white_matter_genes,
multi_gene_method = "z_score",
cap_percentile = 0.95
)
print(p6)
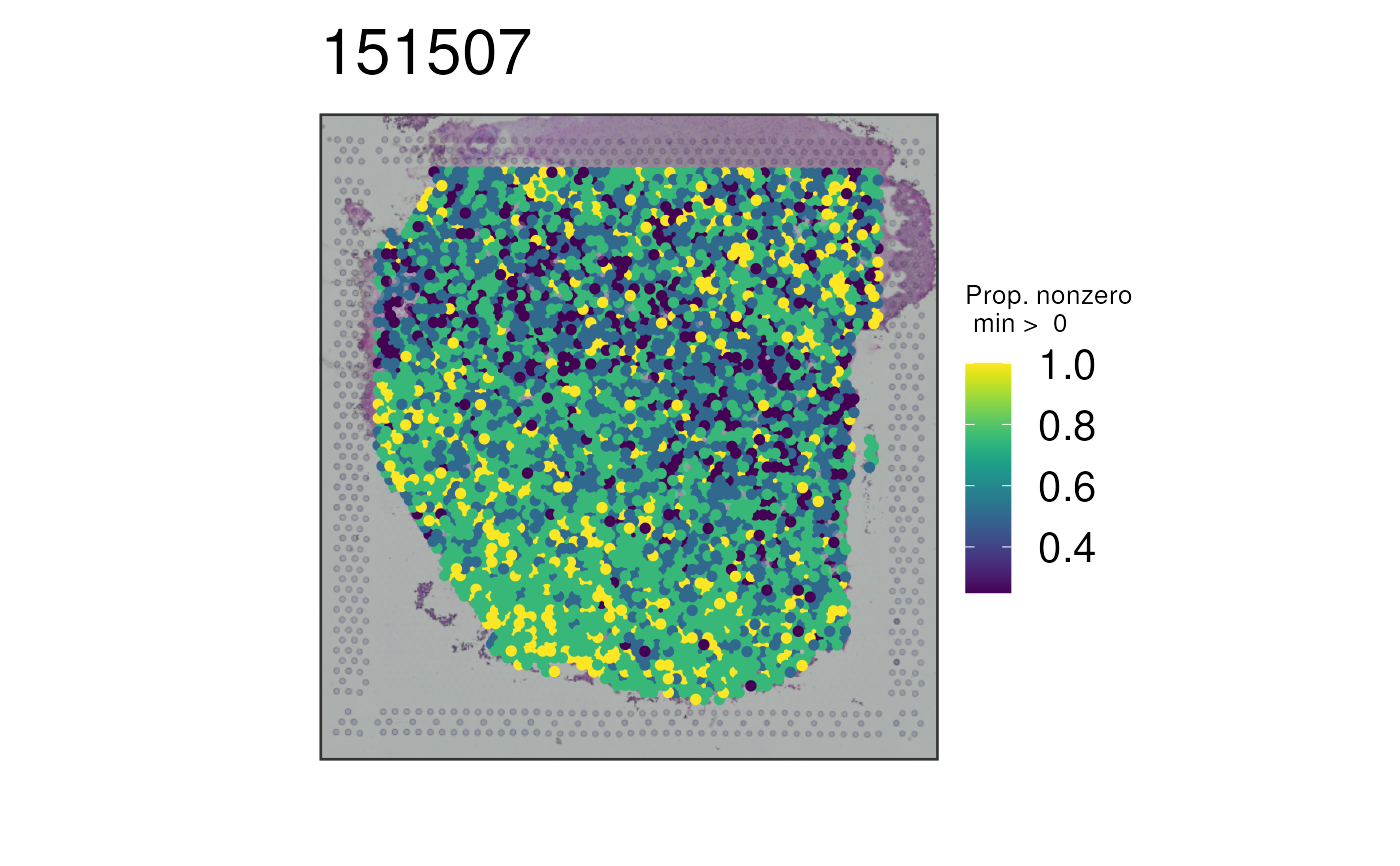
## Plot all white matter markers at once using the sparsity combination
## method
p7 <- vis_gene(
spe = spe,
sampleid = "151507",
geneid = white_matter_genes,
multi_gene_method = "sparsity"
)
print(p7)
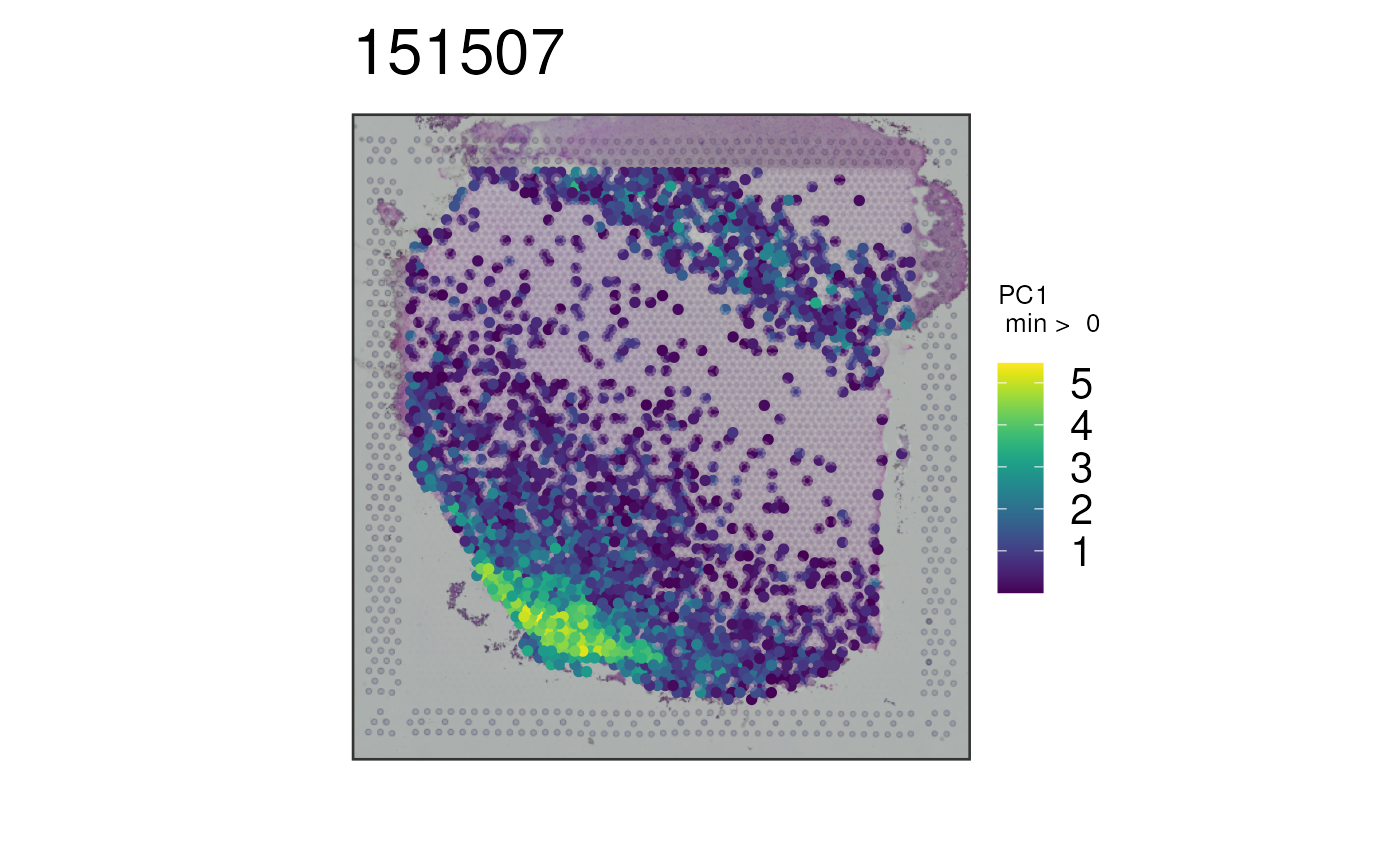
## Plot all white matter markers at once using the PCA combination
## method
p8 <- vis_gene(
spe = spe,
sampleid = "151507",
geneid = white_matter_genes,
multi_gene_method = "pca"
)
print(p8)
}
#> 2026-03-27 00:07:51.447235 loading file /github/home/.cache/R/BiocFileCache/1a6c5ad86da_Human_DLPFC_Visium_processedData_sce_scran_spatialLIBD.Rdata%3Fdl%3D1