This function visualizes the clusters for one given sample at the spot-level
using (by default) the histology information on the background. This is the
function that does all the plotting behind vis_clus(). To visualize
gene-level (or any continuous variable) use vis_gene_p().
vis_clus_p(
spe,
d,
clustervar,
sampleid = unique(spe$sample_id)[1],
colors,
spatial,
title,
image_id = "lowres",
alpha = NA,
point_size = 2,
auto_crop = TRUE,
na_color = "#CCCCCC40"
)Arguments
- spe
A SpatialExperiment-class object. See
fetch_data()for how to download some example objects orread10xVisiumWrapper()to read inspaceranger --countoutput files and build your ownspeobject.- d
A
data.frame()with the sample-level information. This is typically obtained usingcbind(colData(spe), spatialCoords(spe)).- clustervar
A
character(1)with the name of thecolData(spe)column that has the cluster values.- sampleid
A
character(1)specifying which sample to plot fromcolData(spe)$sample_id(formerlycolData(spe)$sample_name).- colors
A vector of colors to use for visualizing the clusters from
clustervar. If the vector has names, then those should match the values ofclustervar.- spatial
A
logical(1)indicating whether to include the histology layer fromgeom_spatial(). If you plan to use ggplotly() then it's best to set this toFALSE.- title
The title for the plot.
- image_id
A
character(1)with the name of the image ID you want to use in the background.- alpha
A
numeric(1)in the[0, 1]range that specifies the transparency level of the data on the spots.- point_size
A
numeric(1)specifying the size of the points. Defaults to1.25. Some colors look better if you use2for instance.- auto_crop
A
logical(1)indicating whether to automatically crop the image / plotting area, which is useful if the Visium capture area is not centered on the image and if the image is not a square.- na_color
A
character(1)specifying a color for the NA values. If you setalpha = NAthen it's best to setna_colorto a color that has alpha blending already, which will make non-NA values pop up more and the NA values will show with a lighter color. This behavior is lost whenalphais set to a non-NAvalue.
Value
A ggplot2 object.
See also
Other Spatial cluster visualization functions:
frame_limits(),
vis_clus(),
vis_grid_clus(),
vis_image()
Examples
if (enough_ram()) {
## Obtain the necessary data
if (!exists("spe")) spe <- fetch_data("spe")
spe_sub <- spe[, spe$sample_id == "151673"]
## Use the manual color palette by Lukas M Weber
## Don't plot the histology information
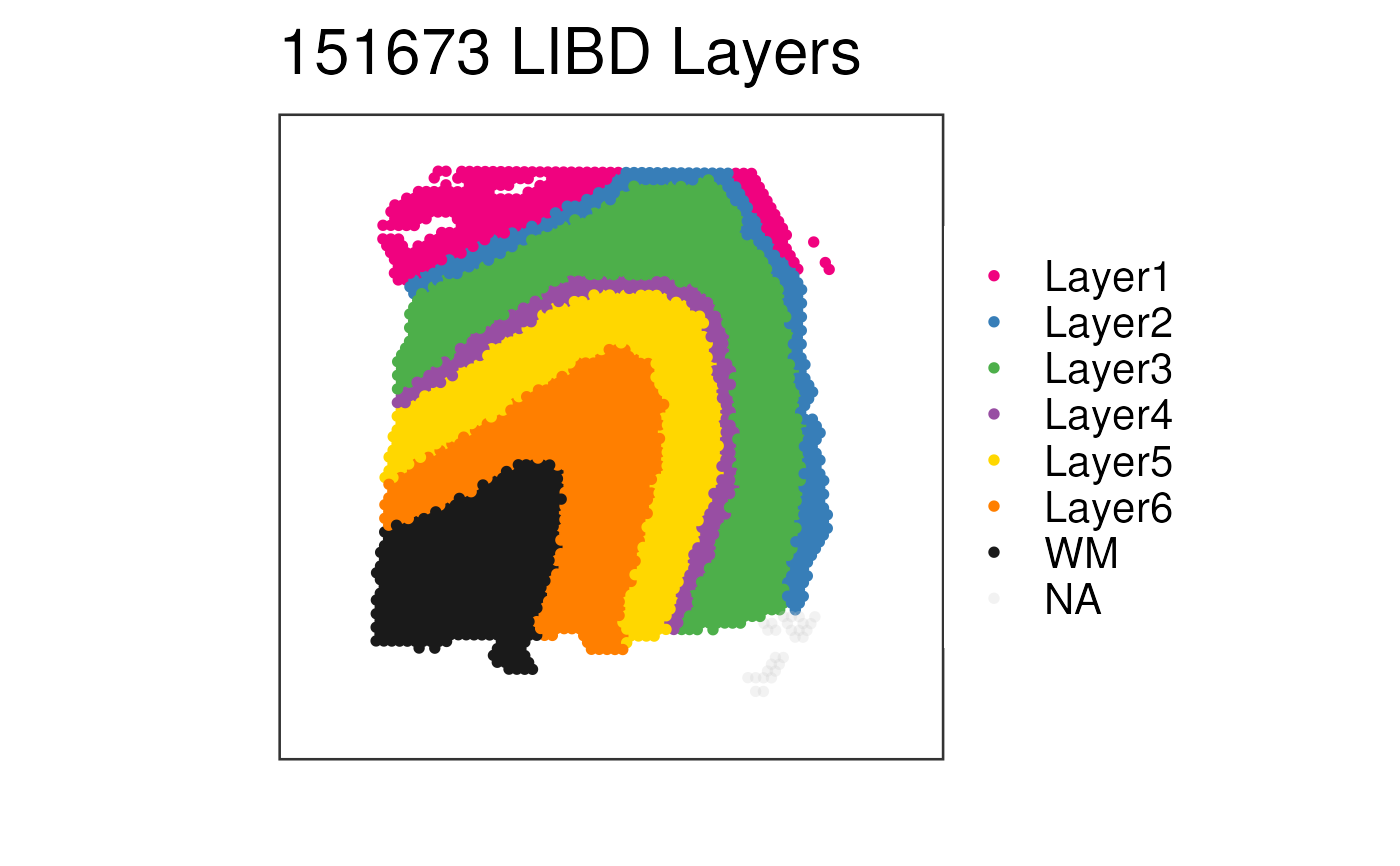
p <- vis_clus_p(
spe = spe_sub,
d = as.data.frame(cbind(colData(spe_sub), SpatialExperiment::spatialCoords(spe_sub)), optional = TRUE),
clustervar = "layer_guess_reordered",
sampleid = "151673",
colors = libd_layer_colors,
title = "151673 LIBD Layers",
spatial = FALSE
)
print(p)
## Clean up
rm(spe_sub)
}
#> 2026-01-09 17:23:58.15862 loading file /github/home/.cache/R/BiocFileCache/1b5c76453a2f_Human_DLPFC_Visium_processedData_sce_scran_spatialLIBD.Rdata%3Fdl%3D1